

Microbial uptake by the respiratory epithelium: outcomes for host and pathogen
- PMID: 30657899
- PMCID: PMC6435450
- DOI: 10.1093/femsre/fuy045
Microbial uptake by the respiratory epithelium: outcomes for host and pathogen
Abstract
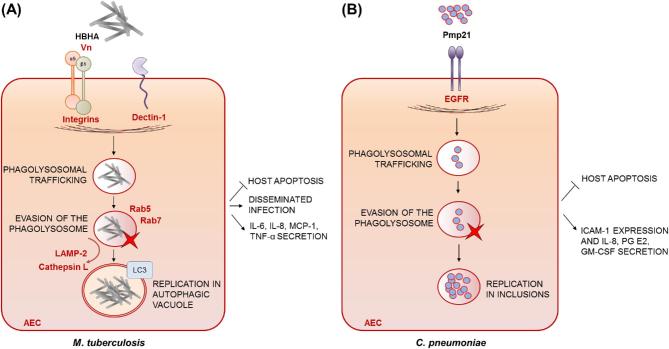
Intracellular occupancy of the respiratory epithelium is a useful pathogenic strategy facilitating microbial replication and evasion of professional phagocytes or circulating antimicrobial drugs. A less appreciated but growing body of evidence indicates that the airway epithelium also plays a crucial role in host defence against inhaled pathogens, by promoting ingestion and quelling of microorganisms, processes that become subverted to favour pathogen activities and promote respiratory disease. To achieve a deeper understanding of beneficial and deleterious activities of respiratory epithelia during antimicrobial defence, we have comprehensively surveyed all current knowledge on airway epithelial uptake of bacterial and fungal pathogens. We find that microbial uptake by airway epithelial cells (AECs) is a common feature of respiratory host-microbe interactions whose stepwise execution, and impacts upon the host, vary by pathogen. Amidst the diversity of underlying mechanisms and disease outcomes, we identify four key infection scenarios and use best-characterised host-pathogen interactions as prototypical examples of each. The emergent view is one in which effi-ciency of AEC-mediated pathogen clearance correlates directly with severity of disease outcome, therefore highlighting an important unmet need to broaden our understanding of the antimicrobial properties of respiratory epithelia and associated drivers of pathogen entry and intracellular fate.
Keywords: airway epithelial cells (AECs); epithelial responses; microbial uptake; microbicidal activities; pathogenesis; respiratory epithelium.
© Crown copyright 2019.
Figures




References
-
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 1999;17:593–623. - PubMed
-
- Agarwal V, Ahl J, Riesbeck K et al.. An alternative role of C1q in bacterial infections: facilitating Streptococcus pneumoniae adherence and invasion of host cells. J Immunol 2013a;191:4235–45. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
