

Epithelial Mutations in Endometriosis: Link to Ovarian Cancer
- PMID: 30657901
- PMCID: PMC6382454
- DOI: 10.1210/en.2018-00794
Epithelial Mutations in Endometriosis: Link to Ovarian Cancer
Abstract
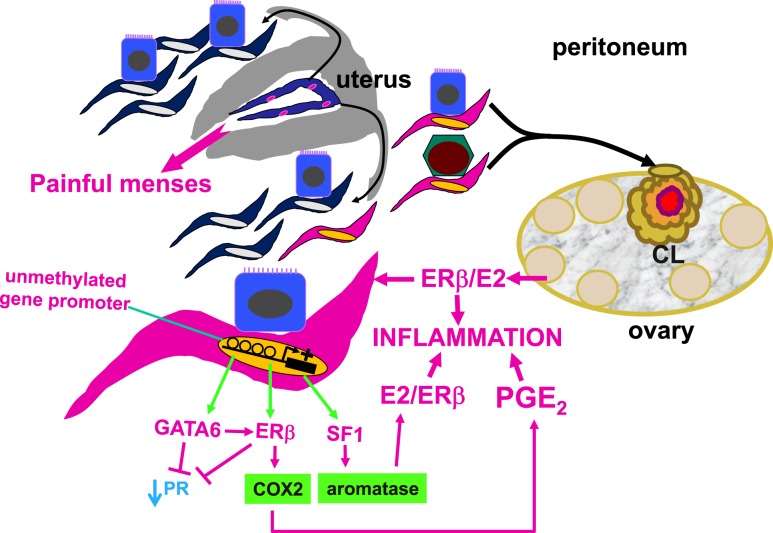
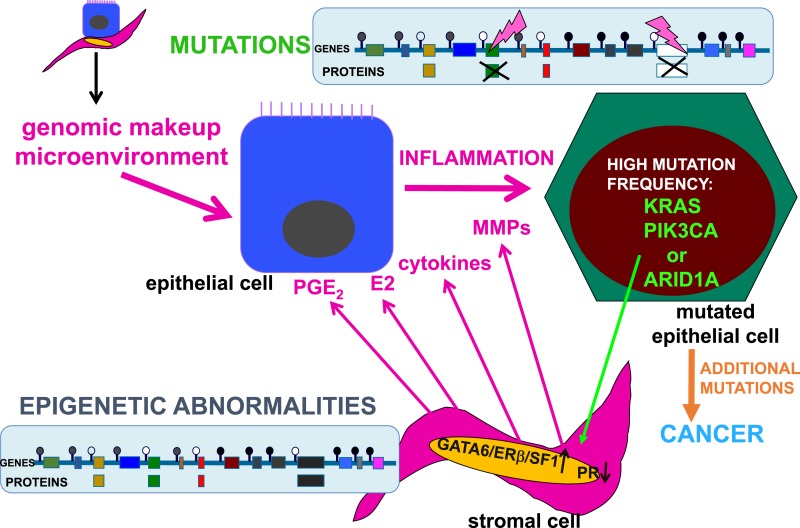
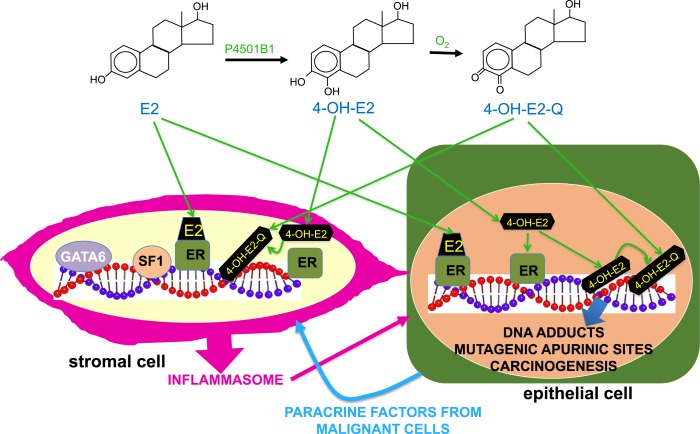
Epidemiologic and histopathologic associations between endometriosis and epithelial ovarian cancer have been reported; however, the underlying molecular and cellular mechanisms are not well understood. A possible genetic link has been suggested in recent publications. Driver mutations in PIK3CA, KRAS, ARID1A, and other genes have been found in the epithelium of intrauterine endometrial tissue, ovarian and extraovarian pelvic endometriosis tissue, ovarian cancers associated with endometriosis (i.e., clear cell and endometrioid type), and other epithelial ovarian cancers. This makes sense because pelvic endometriosis occurs primarily as a result of retrograde menstruation and implantation of endometrial tissue fragments in ovarian inclusion cysts or extraovarian peritoneal or subperitoneal sites. Unlike epithelial cells, endometriotic stromal cells are mutation free but contain widespread epigenetic defects that alter gene expression and induce a progesterone-resistant and intensely inflammatory environment, driven by estrogen via estrogen receptor-β. The resulting increased estrogenic action in the stroma drives inflammation and sends paracrine signals to neighboring epithelial cells to enhance proliferation. In addition, massively high concentrations of estrogen in the ovary may exert an additional and direct genotoxic effect on DNA and cause accumulation of additional mutations and malignant transformation in initially mutated endometriotic epithelial cells in an ovarian endometrioma, which may initiate epithelial ovarian cancer. The same epithelial mutations and inflammatory processes in stroma are seen in extraovarian deep-infiltrating endometriosis, but carcinogenesis does not occur. We provide a focused review of the literature and discuss the implications of recent genetic breakthroughs linking endometriosis and ovarian cancer.
Copyright © 2019 Endocrine Society.
Figures
References
-
- Bulun SE. Endometriosis In: Strauss J, Narnieri R, eds. Yen & Jaffe’s Reproductive Endocrinology. Philadelphia, PA: Elsevier; 2018:609–642.
-
- Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noë M, Horlings HM, Lum A, Jones S, Senz J, Seckin T, Ho J, Wu RC, Lac V, Ogawa H, Tessier-Cloutier B, Alhassan R, Wang A, Wang Y, Cohen JD, Wong F, Hasanovic A, Orr N, Zhang M, Popoli M, McMahon W, Wood LD, Mattox A, Allaire C, Segars J, Williams C, Tomasetti C, Boyd N, Kinzler KW, Gilks CB, Diaz L, Wang TL, Vogelstein B, Yong PJ, Huntsman DG, Shih IM. Cancer-associated mutations in endometriosis without cancer. N Engl J Med. 2017;376(19):1835–1848. - PMC - PubMed
-
- Li X, Zhang Y, Zhao L, Wang L, Wu Z, Mei Q, Nie J, Li X, Li Y, Fu X, Wang X, Meng Y, Han W. Whole-exome sequencing of endometriosis identifies frequent alterations in genes involved in cell adhesion and chromatin-remodeling complexes. Hum Mol Genet. 2014;23(22):6008–6021. - PubMed
-
- Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, Yamawaki K, Adachi S, Takahashi T, Kase H, Tanaka K, Yamamoto T, Motoyama T, Inoue I, Enomoto T. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Reports. 2018;24(7):1777–1789. - PubMed
-
- Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol. 1927;14(4):422–469.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
