

Interstitial lung disease associated with systemic sclerosis (SSc-ILD)
- PMID: 30658650
- PMCID: PMC6339436
- DOI: 10.1186/s12931-019-0980-7
Interstitial lung disease associated with systemic sclerosis (SSc-ILD)
Abstract
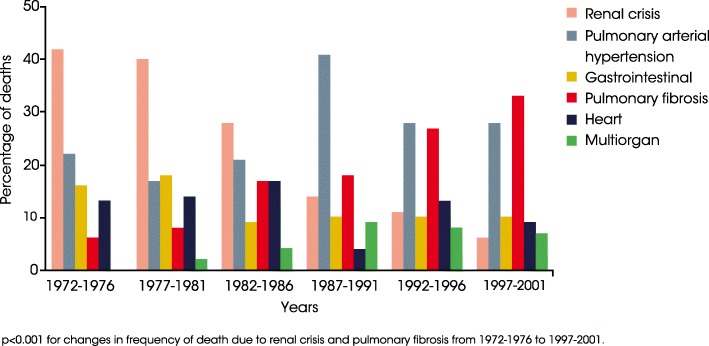
Background: Systemic sclerosis (SSc) is a rare connective tissue disease with a heterogeneous clinical course. Interstitial lung disease (ILD) is a common manifestation of SSc and a leading cause of death.
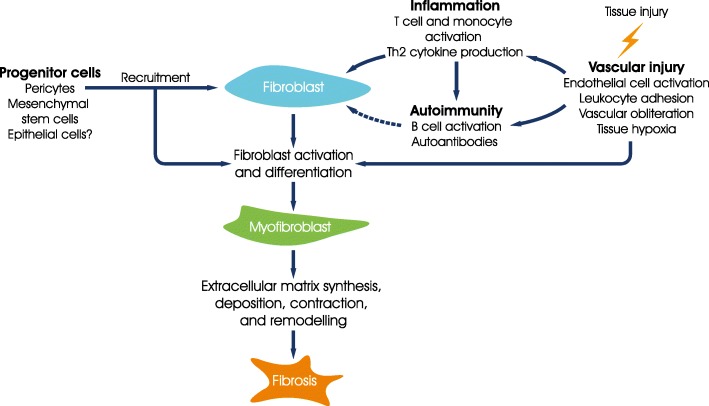
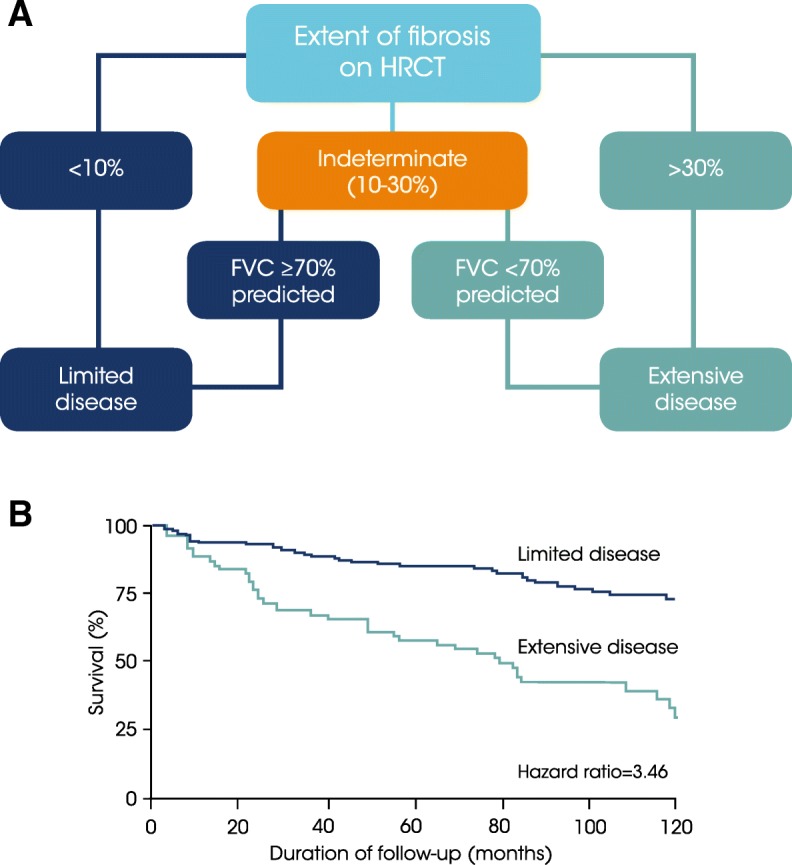
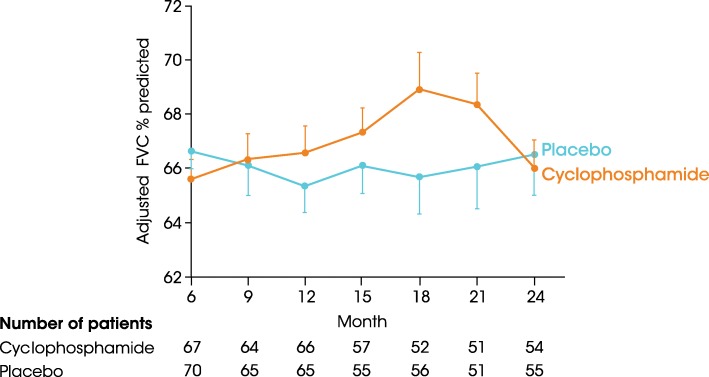
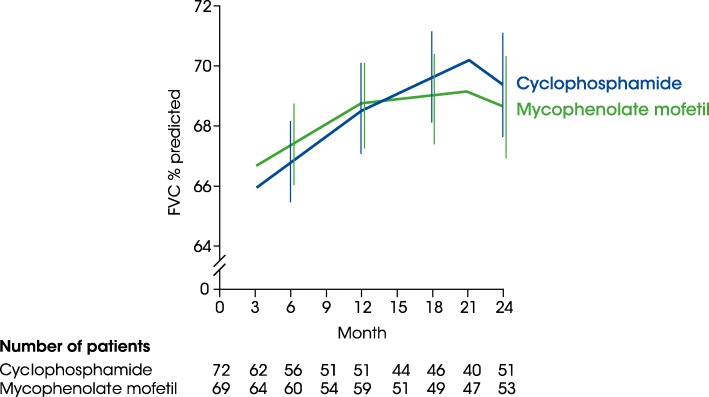
Main body: All patients newly diagnosed with SSc should receive a comprehensive clinical evaluation, including assessment of respiratory symptoms, a high-resolution computed tomography (HRCT) scan of the chest, and pulmonary function tests. ILD can develop in any patient with SSc, including those with pulmonary hypertension, but the risk is increased in those with diffuse (rather than limited) cutaneous SSc, those with anti-Scl-70/anti-topoisomerase I antibody, and in the absence of anti-centromere antibody. While it can occur at any time, the risk of developing ILD is greatest early in the course of SSc, so patients should be monitored closely in the first few years after diagnosis. An increased extent of lung fibrosis on HRCT and a low forced vital capacity (FVC) are predictors of early mortality. While not all patients will require treatment, current approaches to the treatment of progressive SSc-ILD focus on immunosuppressant therapies, including cyclophosphamide and mycophenolate mofetil. In patients with severe and/or rapidly progressive disease, both haematopoietic stem cell transplantation (HSCT) and lung transplantation have been successfully used. A number of medications, including the two drugs approved for the treatment of idiopathic pulmonary fibrosis (IPF), are under active investigation as potential new therapies for SSc-ILD.
Conclusions: Physicians managing patients with SSc should maintain a high level of suspicion and regularly monitor for ILD, particularly in the first few years after diagnosis.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Vincent Cottin has received consultancy fees from Actelion, Bayer, Boehringer Ingelheim, Galapagos, Gilead, Merck Sharp & Dohme, Novartis, Roche, Sanofi; fees for being a member of Data and Safety Monitoring Boards from Celgene, Galapagos, Promedior; and grants paid to his institution from Boehringer Ingelheim and Roche. Kevin Brown has received personal fees from AstraZeneca, Bayer, Biogen Idec, Boehringer Ingelheim, Galapagos, Galecto, Gilead, MedImmune, Novartis, Aeolus, ProMetic, Patara, Third Pole and aTyr, as well as grants from NHLBI.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Wollheim FA. Classification of systemic sclerosis. Visions and reality Rheumatology. 2005;44:1212–1216. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
