

A single discrete Rab5-binding site in phosphoinositide 3-kinase β is required for tumor cell invasion
- PMID: 30659094
- PMCID: PMC6433078
- DOI: 10.1074/jbc.RA118.006032
A single discrete Rab5-binding site in phosphoinositide 3-kinase β is required for tumor cell invasion
Abstract
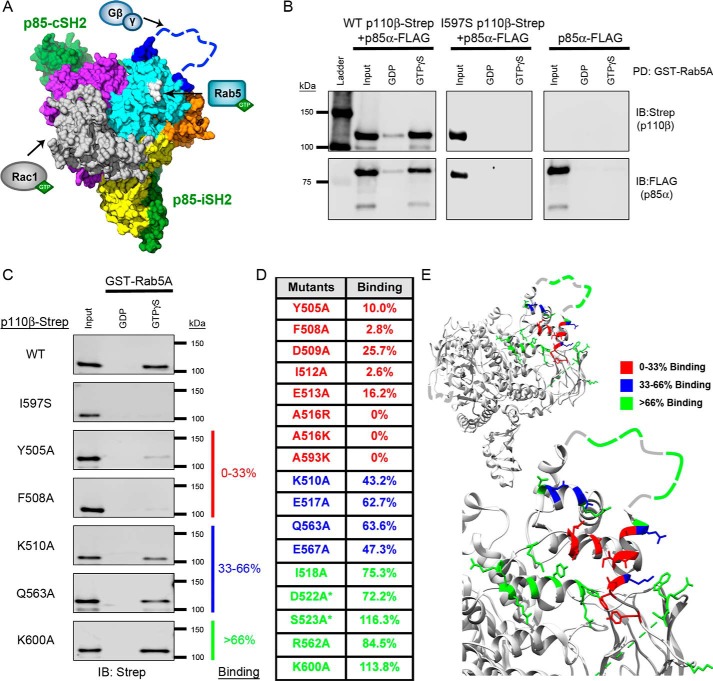
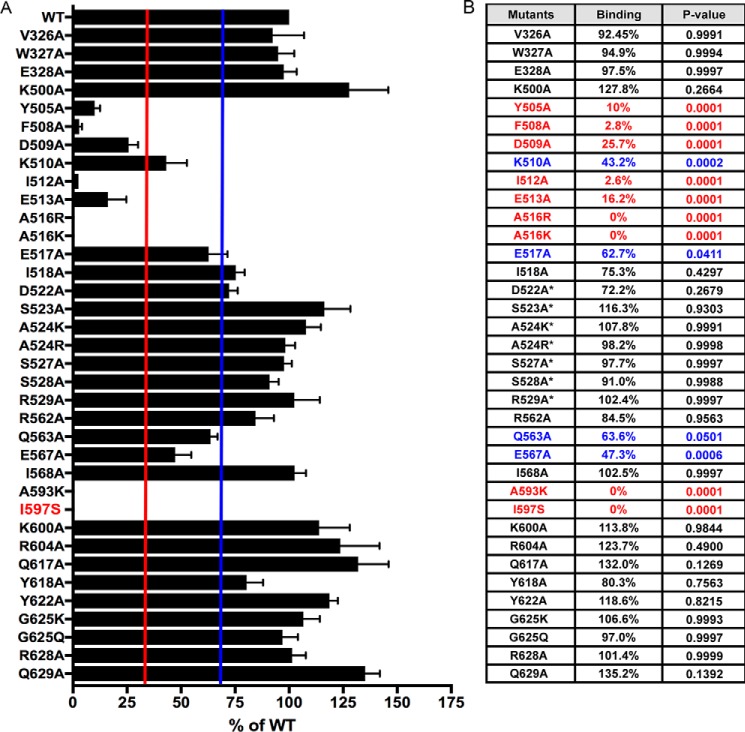
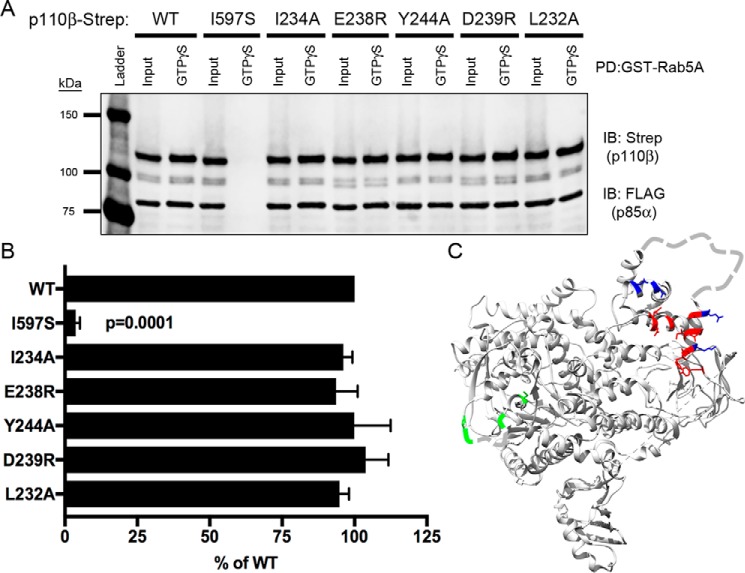
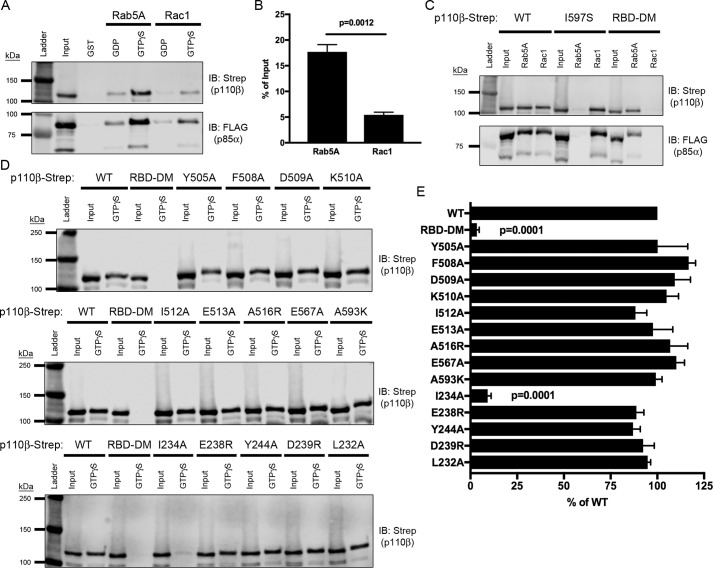
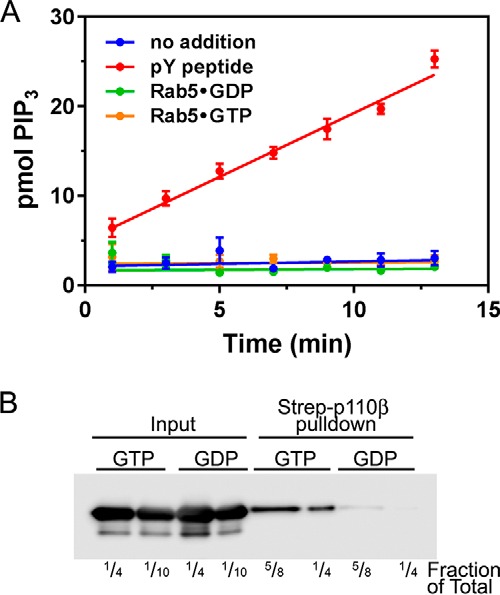
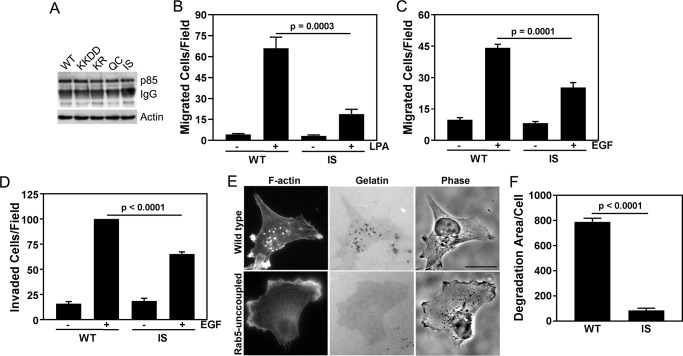
Phosphoinositide 3-kinase β (PI3Kβ) is regulated by receptor tyrosine kinases (RTKs), G protein-coupled receptors (GPCRs), and small GTPases such as Rac1 and Rab5. Our lab previously identified two residues (Gln596 and Ile597) in the helical domain of the catalytic subunit (p110β) of PI3Kβ whose mutation disrupts binding to Rab5. To better define the Rab5-p110β interface, we performed alanine-scanning mutagenesis and analyzed Rab5 binding with an in vitro pulldown assay with GST-Rab5GTP Of the 35 p110β helical domain mutants assayed, 11 disrupted binding to Rab5 without affecting Rac1 binding, basal lipid kinase activity, or Gβγ-stimulated kinase activity. These mutants defined the Rab5-binding interface within p110β as consisting of two perpendicular α-helices in the helical domain that are adjacent to the initially identified Gln596 and Ile597 residues. Analysis of the Rab5-PI3Kβ interaction by hydrogen-deuterium exchange MS identified p110β peptides that overlap with these helices; no interactions were detected between Rab5 and other regions of p110β or p85α. Similarly, the binding of Rab5 to isolated p85α could not be detected, and mutations in the Ras-binding domain (RBD) of p110β had no effect on Rab5 binding. Whereas soluble Rab5 did not affect PI3Kβ activity in vitro, the interaction of these two proteins was critical for chemotaxis, invasion, and gelatin degradation by breast cancer cells. Our results define a single, discrete Rab5-binding site in the p110β helical domain, which may be useful for generating inhibitors to better define the physiological role of Rab5-PI3Kβ coupling in vivo.
Keywords: G protein-coupled receptor (GPCR); GTPase; PIK3CB; PIK3R1; Rab; breast cancer; chemotaxis; invasion; matrix degradation; p85; phosphoinositide 3-kinase (PI 3-kinase); receptor tyrosine kinase.
© 2019 Heitz et al.
Conflict of interest statement
J. M. B. is on the scientific advisory board of Karus Therapeutics
Figures


References
-
- Kurosu H., Maehama T., Okada T., Yamamoto T., Hoshino S., Fukui Y., Ui M., Hazeki O., and Katada T. (1997) Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110β is synergistically activated by the βγ subunits of G proteins and phosphotyrosyl peptide. J. Biol. Chem. 272, 24252–24256 10.1074/jbc.272.39.24252 - DOI - PubMed
-
- Dbouk H. A., Vadas O., Shymanets A., Burke J. E., Salamon R. S., Khalil B. D., Barrett M. O., Waldo G. L., Surve C., Hsueh C., Perisic O., Harteneck C., Shepherd P. R., Harden T. K., Smrcka A. V., et al. (2012) G protein-coupled receptor-mediated activation of p110β by Gβγ is required for cellular transformation and invasiveness. Sci. Signal. 5, ra89 10.1126/scisignal.2003264 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
