

The natural anticancer agent cantharidin alters GPI-anchored protein sorting by targeting Cdc1-mediated remodeling in endoplasmic reticulum
- PMID: 30659098
- PMCID: PMC6422101
- DOI: 10.1074/jbc.RA118.003890
The natural anticancer agent cantharidin alters GPI-anchored protein sorting by targeting Cdc1-mediated remodeling in endoplasmic reticulum
Abstract
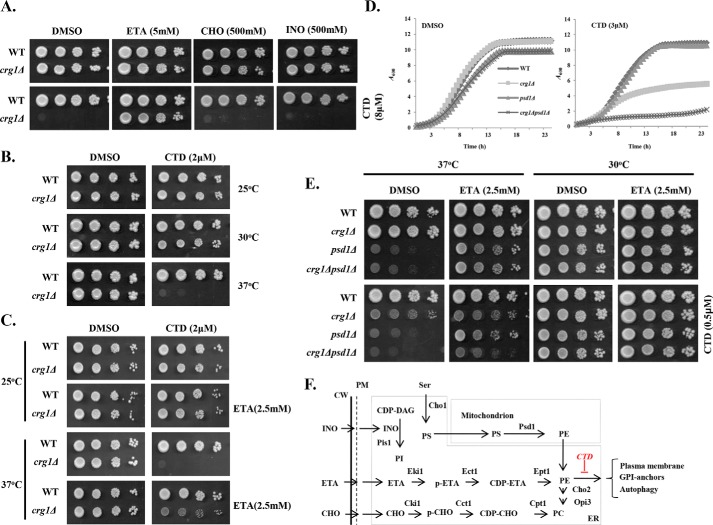
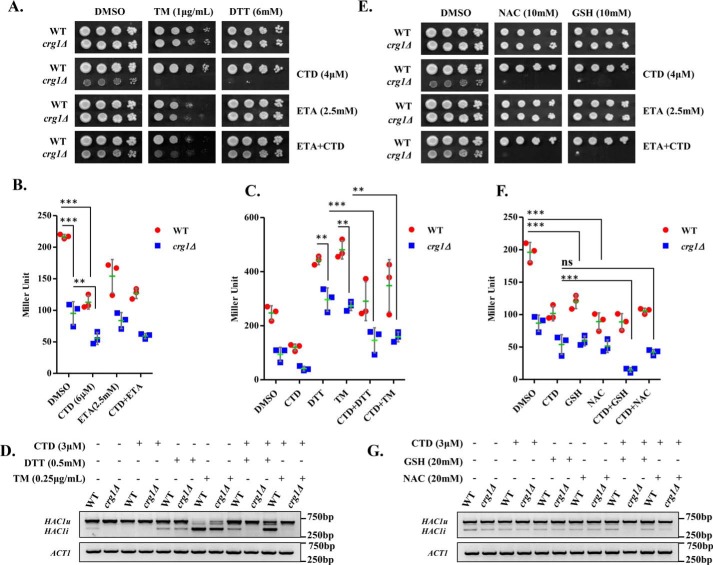
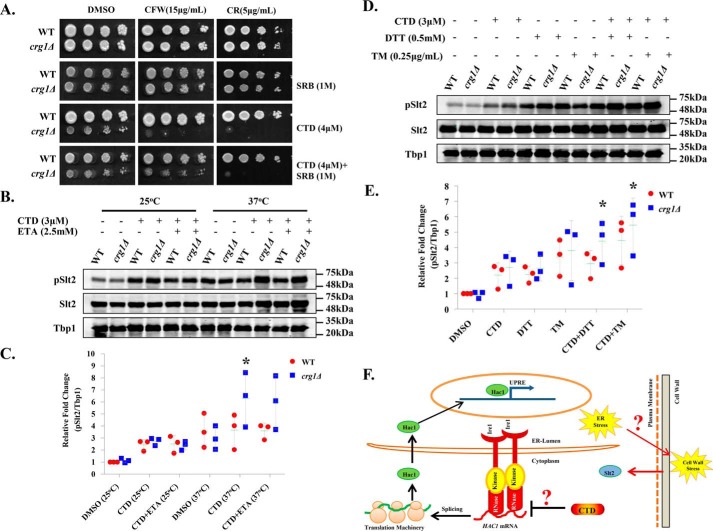
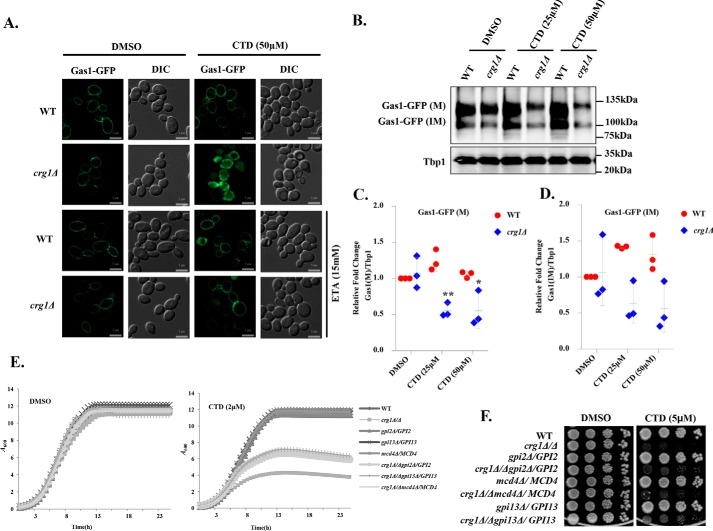
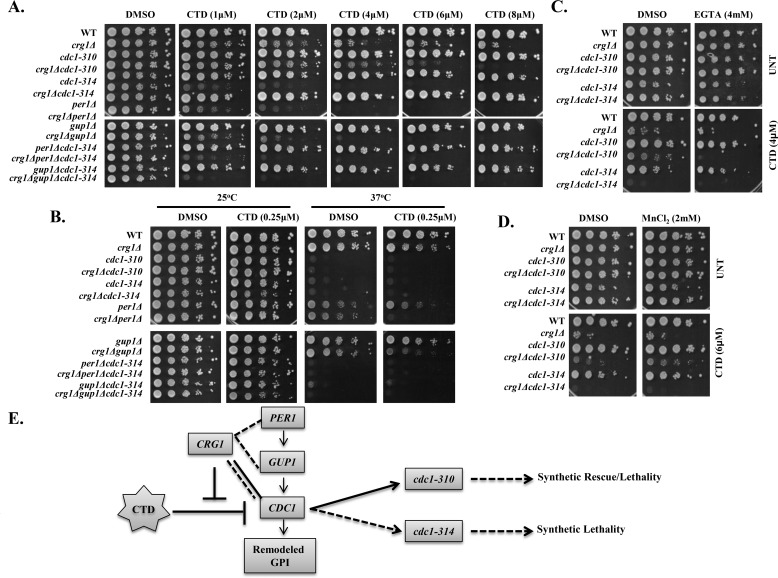
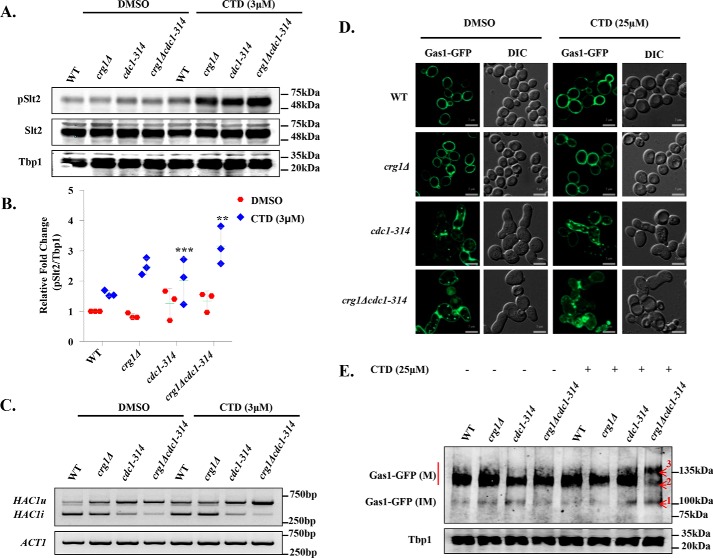
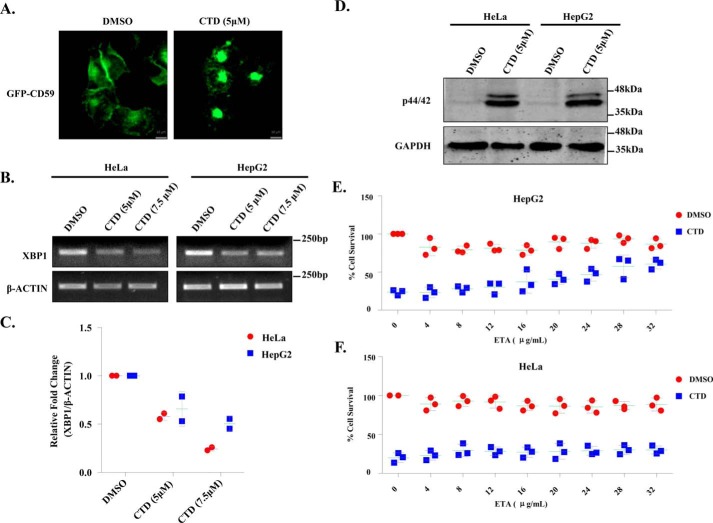
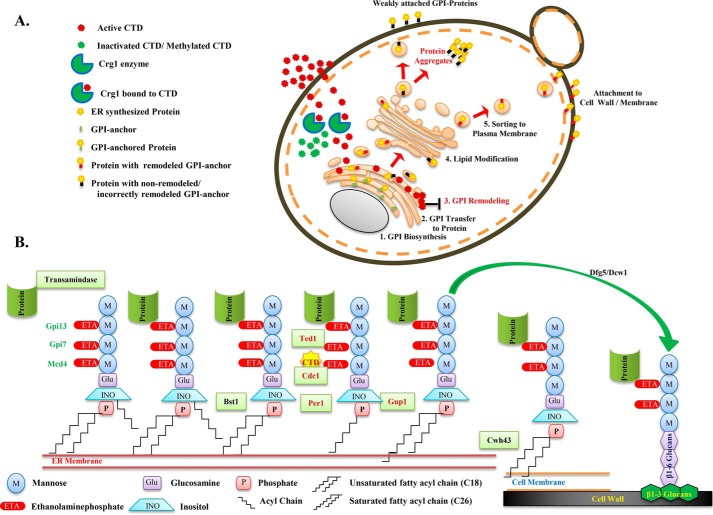
Cantharidin (CTD) is a potent anticancer small molecule produced by several species of blister beetle. It has been a traditional medicine for the management of warts and tumors for many decades. CTD suppresses tumor growth by inducing apoptosis, cell cycle arrest, and DNA damage and inhibits protein phosphatase 2 phosphatase activator (PP2A) and protein phosphatase 1 (PP1). CTD also alters lipid homeostasis, cell wall integrity, endocytosis, adhesion, and invasion in yeast cells. In this study, we identified additional molecular targets of CTD using a Saccharomyces cerevisiae strain that expresses a cantharidin resistance gene (CRG1), encoding a SAM-dependent methyltransferase that methylates and inactivates CTD. We found that CTD specifically affects phosphatidylethanolamine (PE)-associated functions that can be rescued by supplementing the growth media with ethanolamine (ETA). CTD also perturbed endoplasmic reticulum (ER) homeostasis and cell wall integrity by altering the sorting of glycosylphosphatidylinositol (GPI)-anchored proteins. A CTD-dependent genetic interaction profile of CRG1 revealed that the activity of the lipid phosphatase cell division control protein 1 (Cdc1) in GPI-anchor remodeling is the key target of CTD, independently of PP2A and PP1 activities. Moreover, experiments with human cells further suggested that CTD functions through a conserved mechanism in higher eukaryotes. Altogether, we conclude that CTD induces cytotoxicity by targeting Cdc1 activity in GPI-anchor remodeling in the ER.
Keywords: Cdc1; ER homeostasis; ER-Golgi transport; GPI-anchor remodeling; anticancer drug; cantharidin; cell signaling; glycosylphosphatidylinositol (GPI-anchor); lipid metabolism; lipid phosphatase; phospholipid; unfolded protein response (UPR); yeast genetics.
© 2019 Sahu and Tomar.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
Cdc1 removes the ethanolamine phosphate of the first mannose of GPI anchors and thereby facilitates the integration of GPI proteins into the yeast cell wall.Mol Biol Cell. 2014 Nov 1;25(21):3375-88. doi: 10.1091/mbc.E14-06-1033. Epub 2014 Aug 27. Mol Biol Cell. 2014. PMID: 25165136 Free PMC article.
-
The Crg1 N-Terminus Is Essential for Methyltransferase Activity and Cantharidin Resistance in Saccharomyces cerevisiae.Biochemistry. 2019 Apr 2;58(13):1799-1809. doi: 10.1021/acs.biochem.8b01277. Epub 2019 Mar 12. Biochemistry. 2019. PMID: 30830767
-
The yeast p24 complex regulates GPI-anchored protein transport and quality control by monitoring anchor remodeling.Mol Biol Cell. 2011 Aug 15;22(16):2924-36. doi: 10.1091/mbc.E11-04-0294. Epub 2011 Jun 16. Mol Biol Cell. 2011. PMID: 21680708 Free PMC article.
-
Trafficking of glycosylphosphatidylinositol anchored proteins from the endoplasmic reticulum to the cell surface.J Lipid Res. 2016 Mar;57(3):352-60. doi: 10.1194/jlr.R062760. Epub 2015 Oct 8. J Lipid Res. 2016. PMID: 26450970 Free PMC article. Review.
-
GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics.Biochim Biophys Acta. 2012 Aug;1821(8):1050-8. doi: 10.1016/j.bbalip.2012.01.004. Epub 2012 Jan 11. Biochim Biophys Acta. 2012. PMID: 22265715 Review.
Cited by
-
Antitumor potential of the protein phosphatase inhibitor, cantharidin, and selected derivatives.Bioorg Med Chem. 2021 Feb 15;32:116012. doi: 10.1016/j.bmc.2021.116012. Epub 2021 Jan 9. Bioorg Med Chem. 2021. PMID: 33454654 Free PMC article. Review.
-
Potential Physiological Relevance of ERAD to the Biosynthesis of GPI-Anchored Proteins in Yeast.Int J Mol Sci. 2021 Jan 21;22(3):1061. doi: 10.3390/ijms22031061. Int J Mol Sci. 2021. PMID: 33494405 Free PMC article. Review.
-
Sodium cantharidate targets STAT3 and abrogates EGFR inhibitor resistance in osteosarcoma.Aging (Albany NY). 2019 Aug 15;11(15):5848-5863. doi: 10.18632/aging.102193. Epub 2019 Aug 15. Aging (Albany NY). 2019. PMID: 31422383 Free PMC article.
-
Emerging connections between GPI-anchored proteins and their extracellular carriers in colorectal cancer.Extracell Vesicles Circ Nucl Acids. 2023 Jun;4(2):195-217. doi: 10.20517/evcna.2023.17. Epub 2023 May 18. Extracell Vesicles Circ Nucl Acids. 2023. PMID: 37840781 Free PMC article.
-
Tautomycetin Synthetic Analogues: Selective Inhibitors of Protein Phosphatase I.ChemMedChem. 2021 Mar 3;16(5):839-850. doi: 10.1002/cmdc.202000801. Epub 2020 Dec 10. ChemMedChem. 2021. PMID: 33301228 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
