

A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats
- PMID: 30659190
- PMCID: PMC6338754
- DOI: 10.1038/s41467-018-08276-6
A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats
Erratum in
-
Author Correction: A selective inhibitor of mitofusin 1-βIIPKC association improves heart failure outcome in rats.Nat Commun. 2024 Apr 3;15(1):2889. doi: 10.1038/s41467-024-47288-3. Nat Commun. 2024. PMID: 38570501 Free PMC article. No abstract available.
Abstract
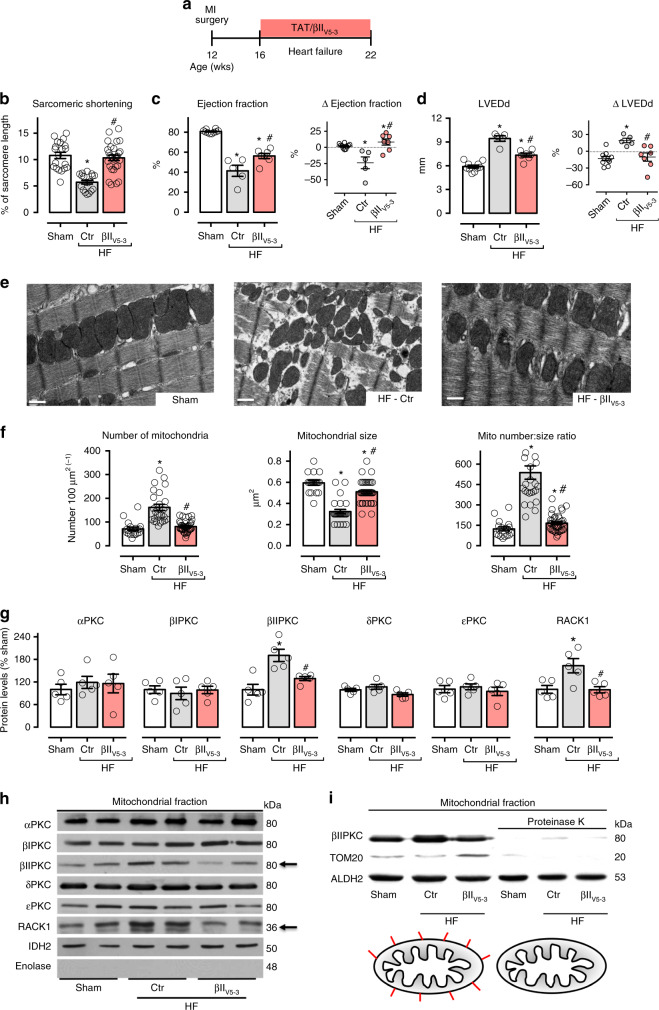
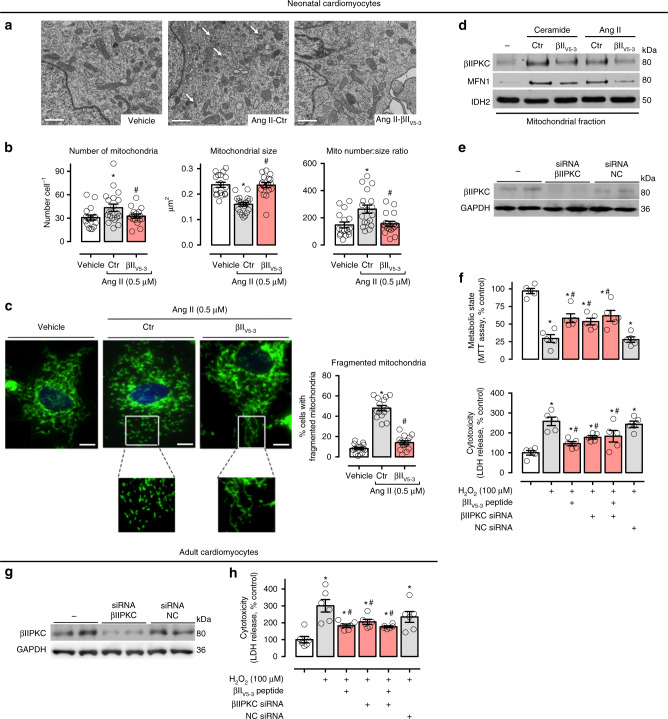
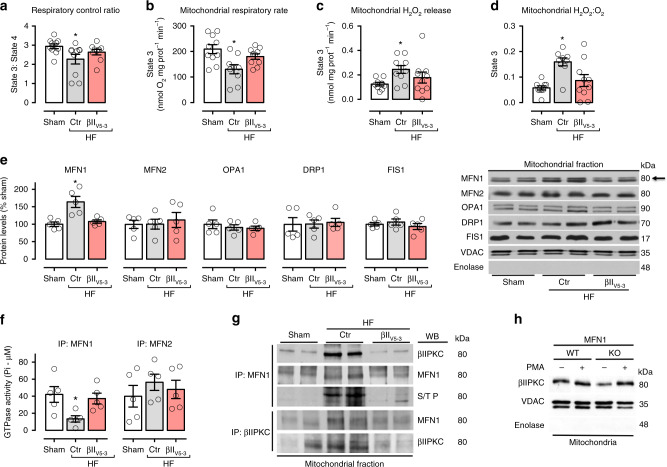
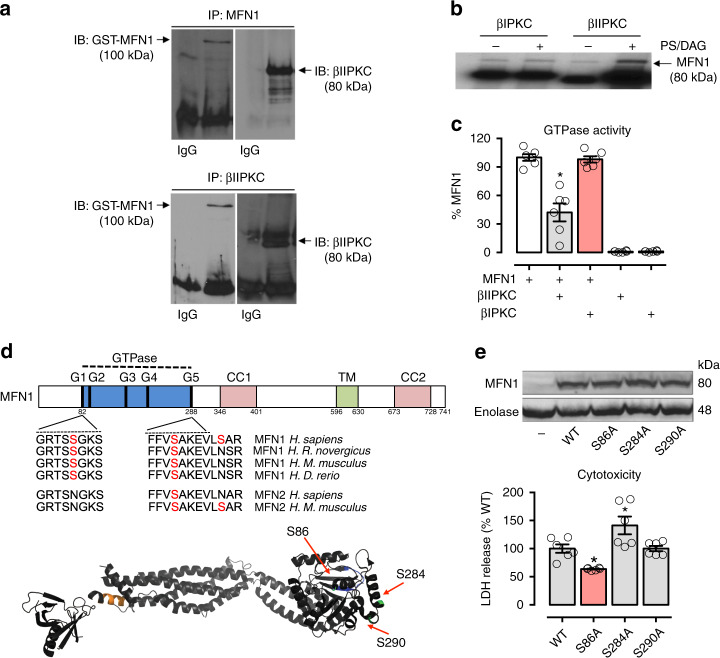
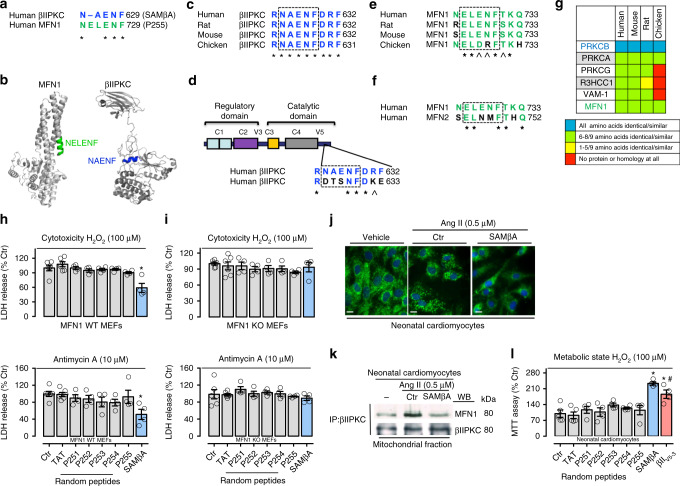
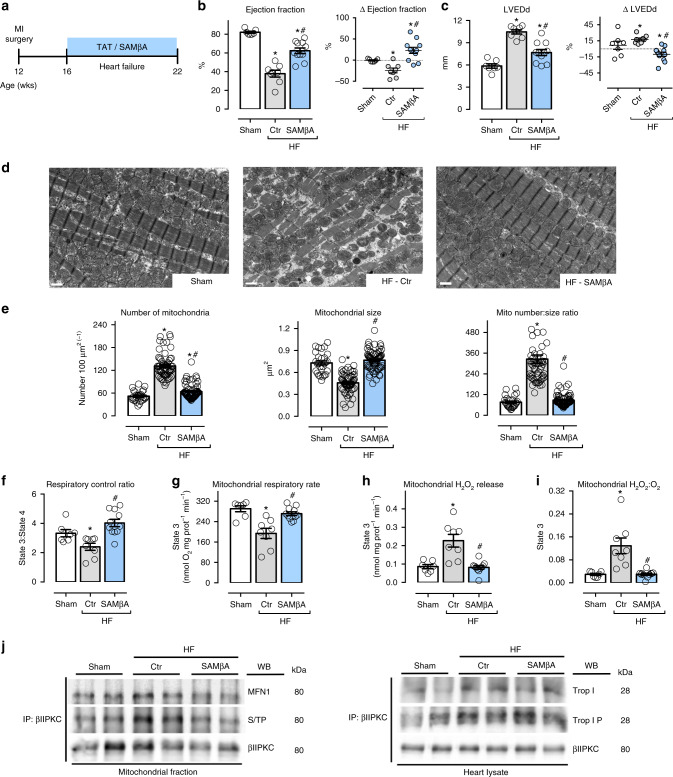
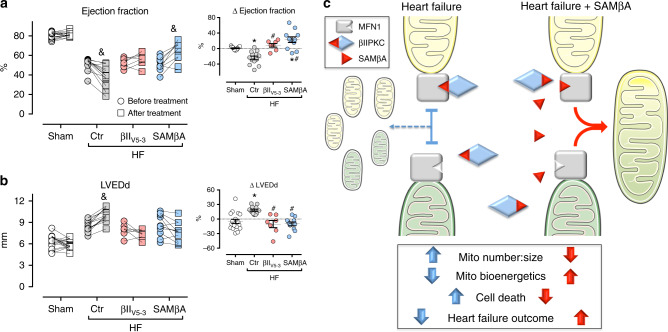
We previously demonstrated that beta II protein kinase C (βIIPKC) activity is elevated in failing hearts and contributes to this pathology. Here we report that βIIPKC accumulates on the mitochondrial outer membrane and phosphorylates mitofusin 1 (Mfn1) at serine 86. Mfn1 phosphorylation results in partial loss of its GTPase activity and in a buildup of fragmented and dysfunctional mitochondria in heart failure. βIIPKC siRNA or a βIIPKC inhibitor mitigates mitochondrial fragmentation and cell death. We confirm that Mfn1-βIIPKC interaction alone is critical in inhibiting mitochondrial function and cardiac myocyte viability using SAMβA, a rationally-designed peptide that selectively antagonizes Mfn1-βIIPKC association. SAMβA treatment protects cultured neonatal and adult cardiac myocytes, but not Mfn1 knockout cells, from stress-induced death. Importantly, SAMβA treatment re-establishes mitochondrial morphology and function and improves cardiac contractility in rats with heart failure, suggesting that SAMβA may be a potential treatment for patients with heart failure.
Conflict of interest statement
J.C.B.F and D.M.-R. are co-inventors of patent on “Antagonists of mitofusin 1 and beta II PKC association for treating heart failure”, PCT/US2019/062854. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
