

Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics
- PMID: 30659192
- PMCID: PMC6338729
- DOI: 10.1038/s41467-018-08191-w
Combining LOPIT with differential ultracentrifugation for high-resolution spatial proteomics
Abstract
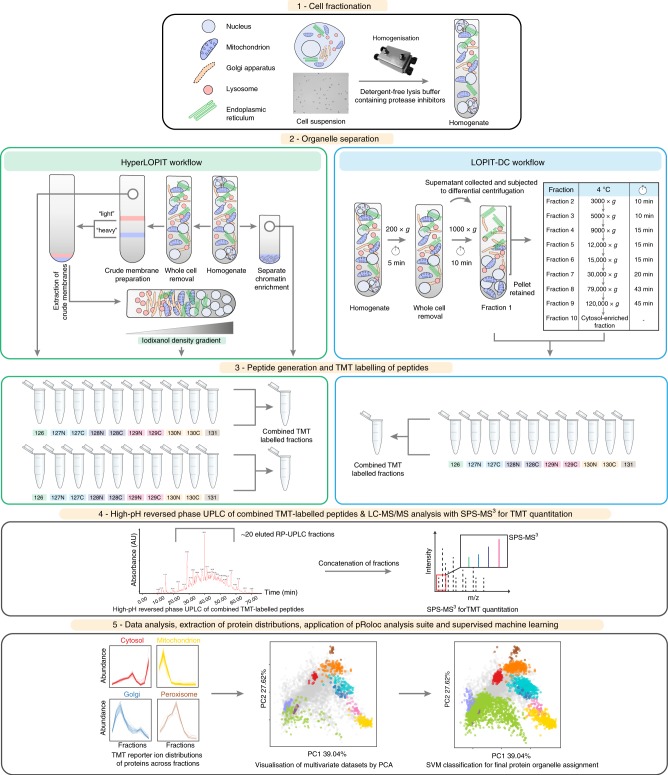
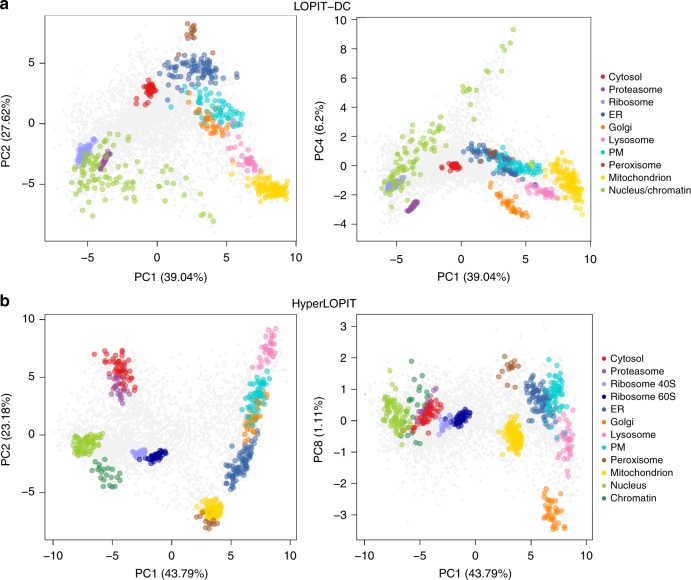
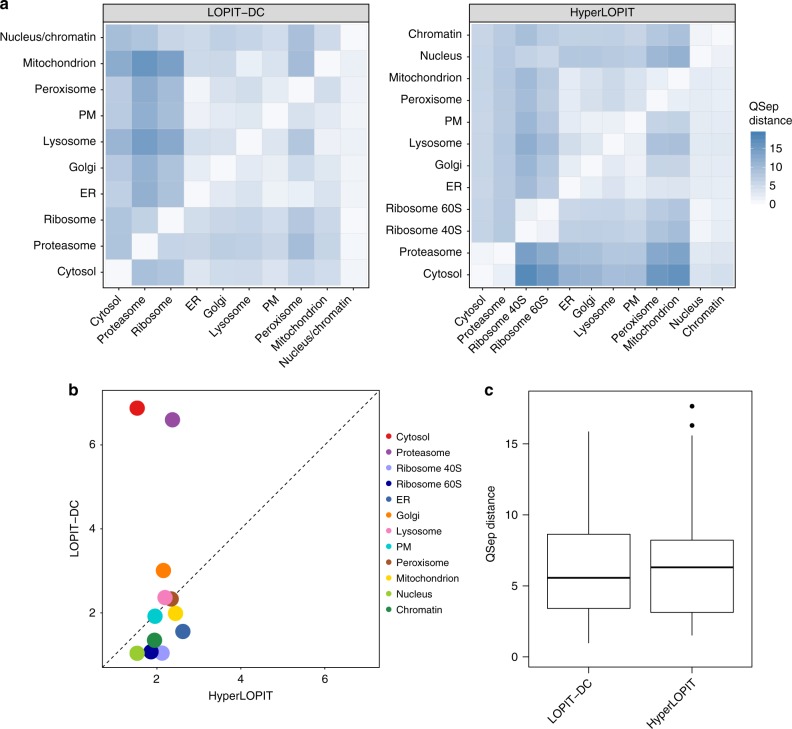
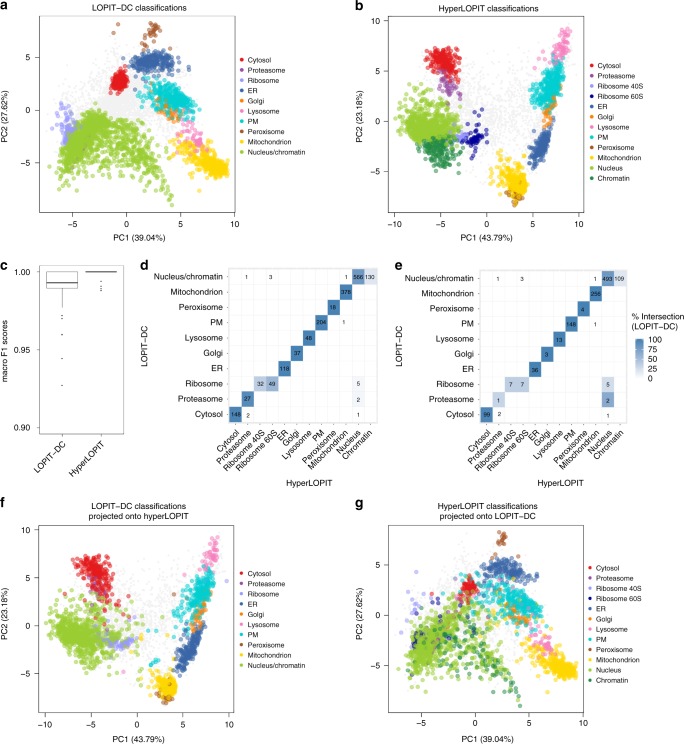
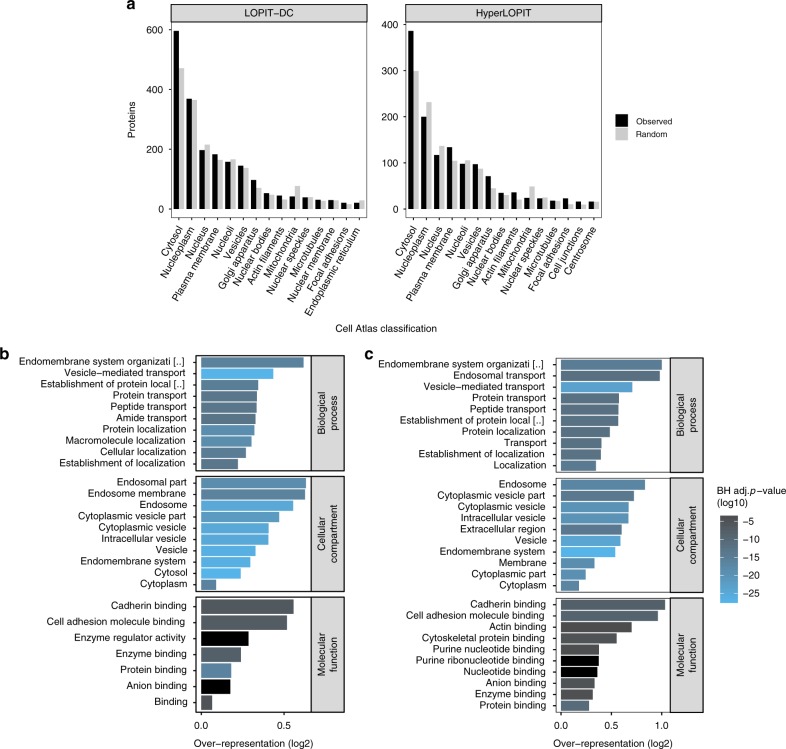
The study of protein localisation has greatly benefited from high-throughput methods utilising cellular fractionation and proteomic profiling. Hyperplexed Localisation of Organelle Proteins by Isotope Tagging (hyperLOPIT) is a well-established method in this area. It achieves high-resolution separation of organelles and subcellular compartments but is relatively time- and resource-intensive. As a simpler alternative, we here develop Localisation of Organelle Proteins by Isotope Tagging after Differential ultraCentrifugation (LOPIT-DC) and compare this method to the density gradient-based hyperLOPIT approach. We confirm that high-resolution maps can be obtained using differential centrifugation down to the suborganellar and protein complex level. HyperLOPIT and LOPIT-DC yield highly similar results, facilitating the identification of isoform-specific localisations and high-confidence localisation assignment for proteins in suborganellar structures, protein complexes and signalling pathways. By combining both approaches, we present a comprehensive high-resolution dataset of human protein localisations and deliver a flexible set of protocols for subcellular proteomics.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
