

Acid sphingomyelinase deficiency protects mitochondria and improves function recovery after brain injury
- PMID: 30662008
- PMCID: PMC6399498
- DOI: 10.1194/jlr.M091132
Acid sphingomyelinase deficiency protects mitochondria and improves function recovery after brain injury
Abstract
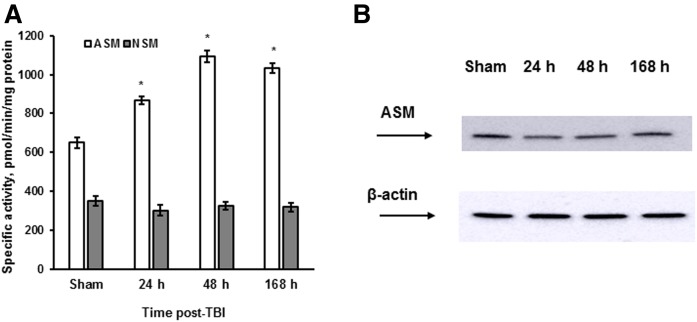
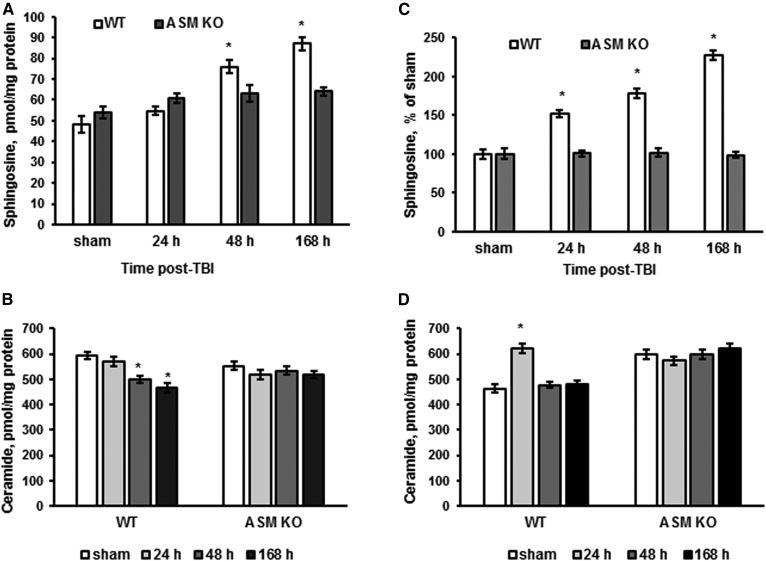
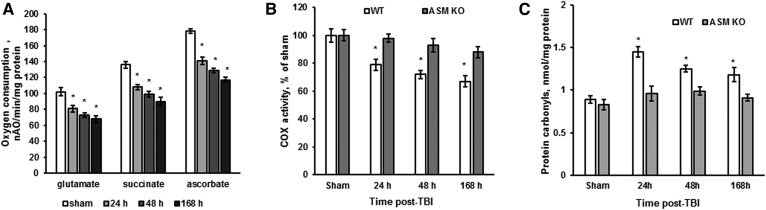
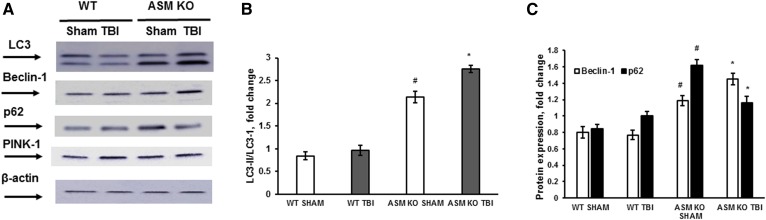
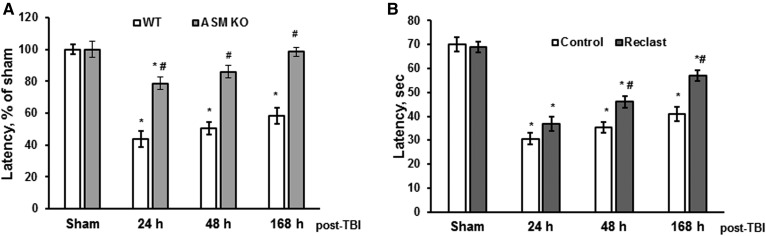
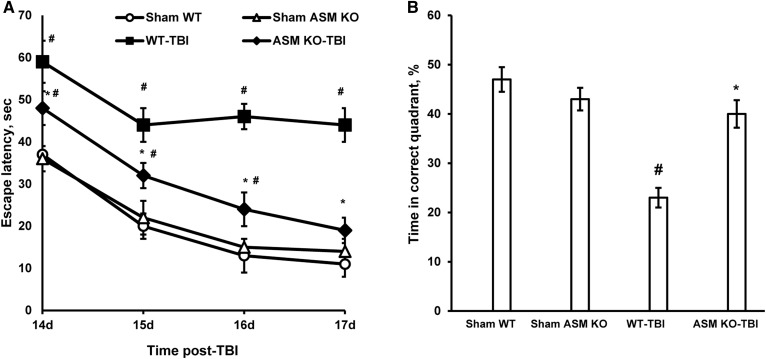
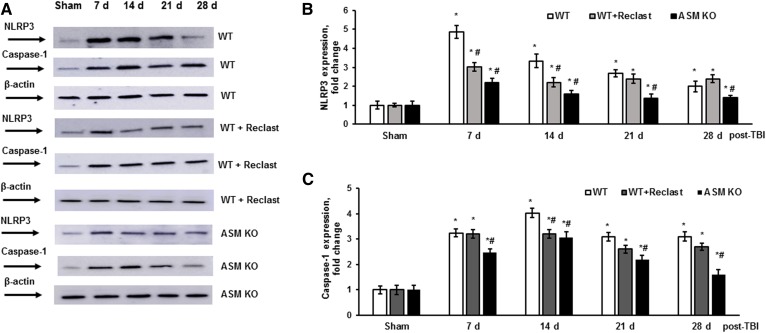
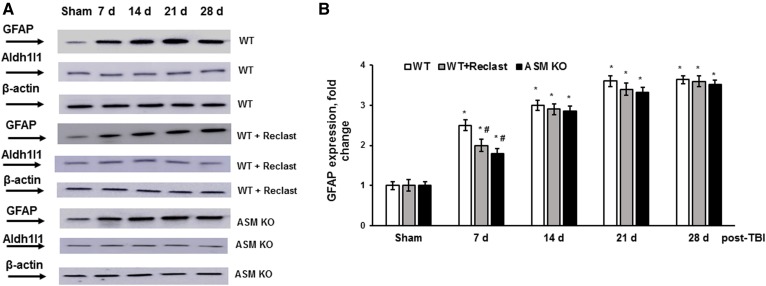
Traumatic brain injury (TBI) is one of the leading causes of disability worldwide and a prominent risk factor for neurodegenerative diseases. The expansion of nervous tissue damage after the initial trauma involves a multifactorial cascade of events, including excitotoxicity, oxidative stress, inflammation, and deregulation of sphingolipid metabolism that further mitochondrial dysfunction and secondary brain damage. Here, we show that a posttranscriptional activation of an acid sphingomyelinase (ASM), a key enzyme of the sphingolipid recycling pathway, resulted in a selective increase of sphingosine in mitochondria during the first week post-TBI that was accompanied by reduced activity of mitochondrial cytochrome oxidase and activation of the Nod-like receptor protein 3 inflammasome. TBI-induced mitochondrial abnormalities were rescued in the brains of ASM KO mice, which demonstrated improved behavioral deficit recovery compared with WT mice. Furthermore, an elevated autophagy in an ASM-deficient brain at the baseline and during the development of secondary brain injury seems to foster the preservation of mitochondria and brain function after TBI. Of note, ASM deficiency attenuated the early stages of reactive astrogliosis progression in an injured brain. These findings highlight the crucial role of ASM in governing mitochondrial dysfunction and brain-function impairment, emphasizing the importance of sphingolipids in the neuroinflammatory response to TBI.
Keywords: astrogliosis; inflammasome; neuroinflammation; sphingolipids.
Figures
Similar articles
-
Instigation of NLRP3 inflammasome activation and glomerular injury in mice on the high fat diet: role of acid sphingomyelinase gene.Oncotarget. 2016 Apr 5;7(14):19031-44. doi: 10.18632/oncotarget.8023. Oncotarget. 2016. PMID: 26980705 Free PMC article.
-
Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury.J Biol Chem. 2014 May 9;289(19):13142-54. doi: 10.1074/jbc.M113.530311. Epub 2014 Mar 21. J Biol Chem. 2014. PMID: 24659784 Free PMC article.
-
Acid Sphingomyelinase Inhibition Mitigates Histopathological and Behavioral Changes in a Murine Model of Traumatic Brain Injury.J Neurotrauma. 2020 Sep 1;37(17):1902-1909. doi: 10.1089/neu.2019.6436. Epub 2020 May 4. J Neurotrauma. 2020. PMID: 32138594 Free PMC article.
-
Traumatic brain injury and mitochondrial dysfunction.Am J Med Sci. 2015 Aug;350(2):132-8. doi: 10.1097/MAJ.0000000000000506. Am J Med Sci. 2015. PMID: 26083647 Review.
-
NLRP3 inflammasome in traumatic brain injury: Its implication in the disease pathophysiology and potential as a therapeutic target.Life Sci. 2023 Feb 1;314:121352. doi: 10.1016/j.lfs.2022.121352. Epub 2022 Dec 30. Life Sci. 2023. PMID: 36592789 Review.
Cited by
-
Crosstalk Between Acid Sphingomyelinase and Inflammasome Signaling and Their Emerging Roles in Tissue Injury and Fibrosis.Front Cell Dev Biol. 2020 Jan 14;7:378. doi: 10.3389/fcell.2019.00378. eCollection 2019. Front Cell Dev Biol. 2020. PMID: 32010692 Free PMC article. Review.
-
Sphingolipid changes in mouse brain and plasma after mild traumatic brain injury at the acute phases.Lipids Health Dis. 2024 Jun 27;23(1):200. doi: 10.1186/s12944-024-02186-x. Lipids Health Dis. 2024. PMID: 38937745 Free PMC article.
-
Brain Energy Metabolism: Astrocytes in Neurodegenerative Diseases.CNS Neurosci Ther. 2023 Jan;29(1):24-36. doi: 10.1111/cns.13982. Epub 2022 Oct 3. CNS Neurosci Ther. 2023. PMID: 36193573 Free PMC article. Review.
-
Systemic Elevation of n-3 Polyunsaturated Fatty Acids (n-3-PUFA) Is Associated with Protection against Visual, Motor, and Emotional Deficits in Mice following Closed-Head Mild Traumatic Brain Injury.Mol Neurobiol. 2021 Nov;58(11):5564-5580. doi: 10.1007/s12035-021-02501-y. Epub 2021 Aug 8. Mol Neurobiol. 2021. PMID: 34365584 Free PMC article.
-
Methylprednisolone Induces Neuro-Protective Effects via the Inhibition of A1 Astrocyte Activation in Traumatic Spinal Cord Injury Mouse Models.Front Neurosci. 2021 May 31;15:628917. doi: 10.3389/fnins.2021.628917. eCollection 2021. Front Neurosci. 2021. PMID: 34135725 Free PMC article.
References
-
- Brooks J. C., Strauss D. J., Shavelle R. M., Paculdo D. R., Hammond F. M., and Harrison-Felix C. L.. 2013. Long-term disability and survival in traumatic brain injury: results from the National Institute on Disability and Rehabilitation Research Model Systems. Arch. Phys. Med. Rehabil. 94: 2203–2209. - PubMed
-
- Maas A. I., Stocchetti N., and Bullock R.. 2008. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 7: 728–741. - PubMed
-
- Werner C., and Engelhard K.. 2007. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 99: 4–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
