

Ginsenoside compound K protects human umbilical vein endothelial cells against oxidized low-density lipoprotein-induced injury via inhibition of nuclear factor-κB, p38, and JNK MAPK pathways
- PMID: 30662298
- PMCID: PMC6323235
- DOI: 10.1016/j.jgr.2017.09.004
Ginsenoside compound K protects human umbilical vein endothelial cells against oxidized low-density lipoprotein-induced injury via inhibition of nuclear factor-κB, p38, and JNK MAPK pathways
Erratum in
-
Corrigendum to "Ginsenoside compound K protects human umbilical vein endothelial cells against oxidized low-density lipoprotein-induced injury via inhibition of nuclear factor-kB, p38, and JNK MAPK pathways" [J Ginseng Res 43 (2019) 95-104].J Ginseng Res. 2024 Nov;48(6):627. doi: 10.1016/j.jgr.2024.09.001. Epub 2024 Sep 2. J Ginseng Res. 2024. PMID: 39583175 Free PMC article.
Abstract
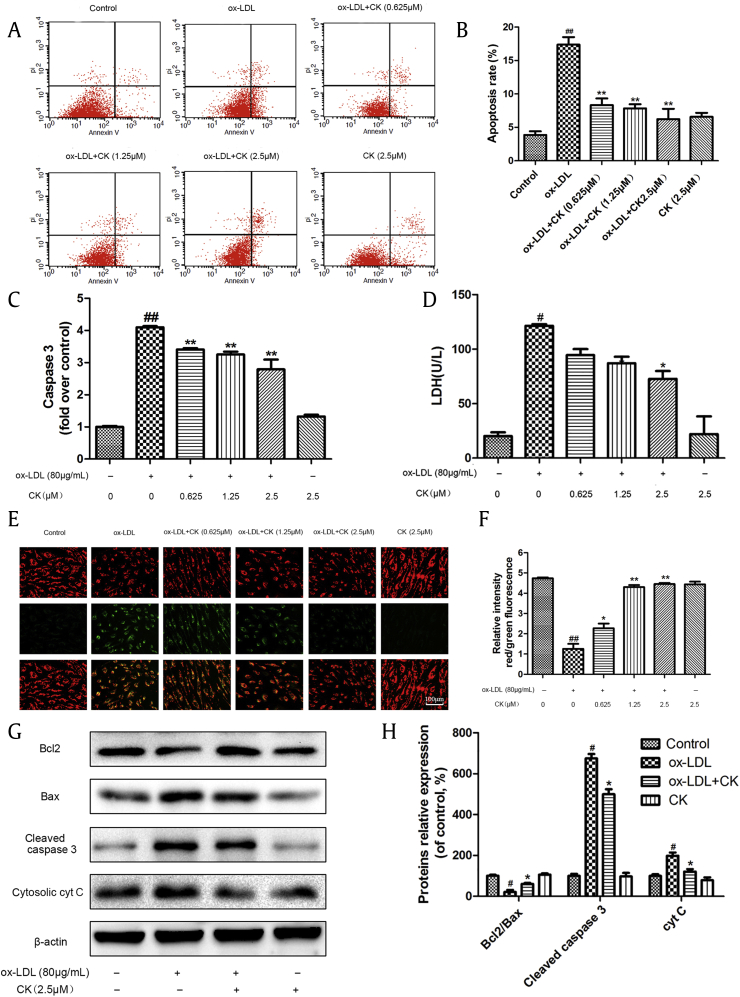
Background: Oxidized low-density lipoprotein (ox-LDL) causes vascular endothelial cell inflammatory response and apoptosis and plays an important role in the development and progression of atherosclerosis. Ginsenoside compound K (CK), a metabolite produced by the hydrolysis of ginsenoside Rb1, possesses strong anti-inflammatory effects. However, whether or not CK protects ox-LDL-damaged endothelial cells and the potential mechanisms have not been elucidated.
Methods: In our study, cell viability was tested using a 3-(4, 5-dimethylthiazol-2yl-)-2,5-diphenyl tetrazolium bromide (MTT) assay. Expression levels of interleukin-6, monocyte chemoattractant protein-1, tumor necrosis factor-α, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 were determined by enzyme-linked immunosorbent assay and Western blotting. Mitochondrial membrane potential (ΔΨm) was detected using JC-1. The cell apoptotic percentage was measured by the Annexin V/ propidium iodide (PI) assay, lactate dehydrogenase, and caspase-3 expression. Apoptosis-related proteins, nuclear factor (NF)-κB, and mitogen-activated protein kinases (MAPK) signaling pathways protein expression were quantified by Western blotting.
Results: Our results demonstrated that CK could ameliorate ox-LDL-induced human umbilical vein endothelial cells (HUVECs) inflammation and apoptosis, NF-κB nuclear translocation, and the phosphorylation of p38 and c-Jun N-terminal kinase (JNK). Moreover, anisomycin, an activator of p38 and JNK, significantly abolished the anti-apoptotic effects of CK.
Conclusion: These results demonstrate that CK prevents ox-LDL-induced HUVECs inflammation and apoptosis through inhibiting the NF-κB, p38, and JNK MAPK signaling pathways. Thus, CK is a candidate drug for atherosclerosis treatment.
Keywords: apoptosis; ginsenoside compound K; human umbilical vein endothelial cells; inflammation; oxidized low-density lipoprotein.
Figures






References
-
- Siegel D., Devaraj S., Mitra A., Raychaudhuri S.P., Raychaudhuri S.K., Jialal I. Inflammation, atherosclerosis, and psoriasis. Clin Rev Allergy Immunol. 2013;44:194–204. - PubMed
-
- Chistiakov D.A., Orekhov A.N., Bobryshev Y.V. LOX-1-mediated effects on vascular cells in atherosclerosis. Cell Physiol Biochem. 2016;38:1851–1859. - PubMed
-
- Ogura S., Kakino A., Sato Y., Fujita Y., Iwamoto S., Otsui K., Yoshimoto R., Sawamura T. Lox-1: the multifunctional receptor underlying cardiovascular dysfunction. Circ J. 2009;73:1993–1999. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
