

HomoplasyFinder: a simple tool to identify homoplasies on a phylogeny
- PMID: 30663960
- PMCID: PMC6412054
- DOI: 10.1099/mgen.0.000245
HomoplasyFinder: a simple tool to identify homoplasies on a phylogeny
Abstract
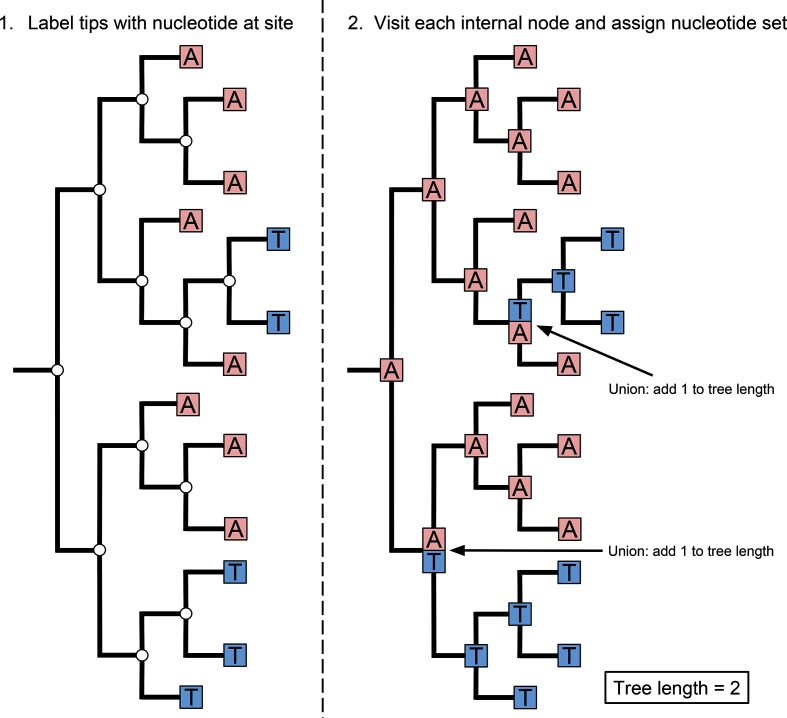
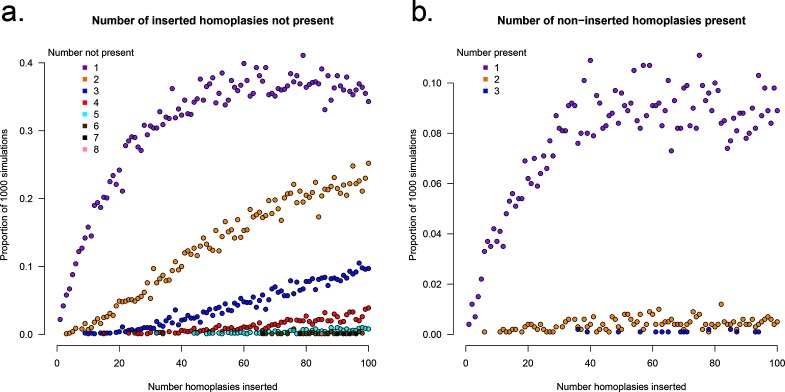
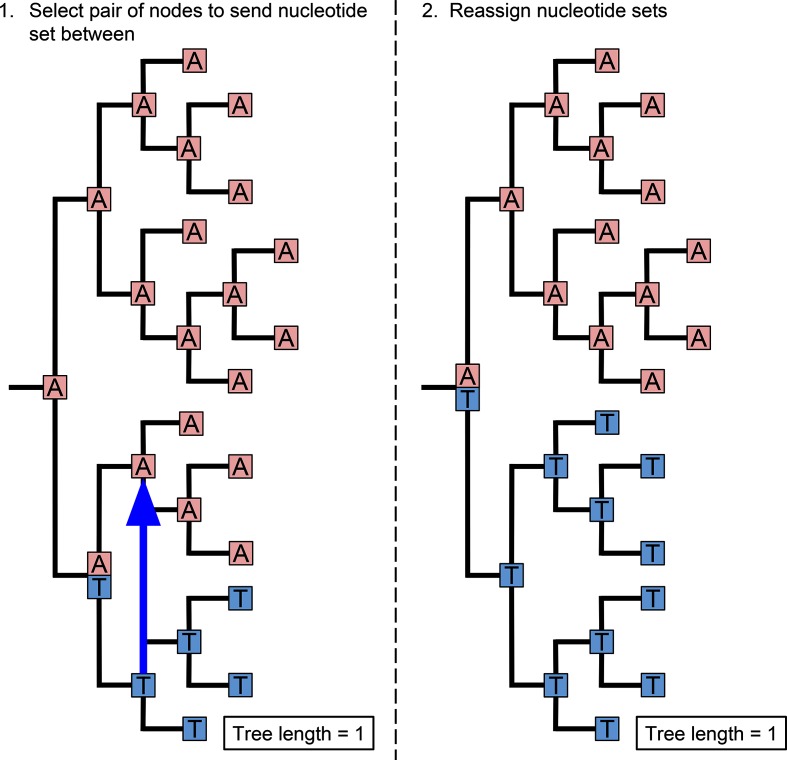
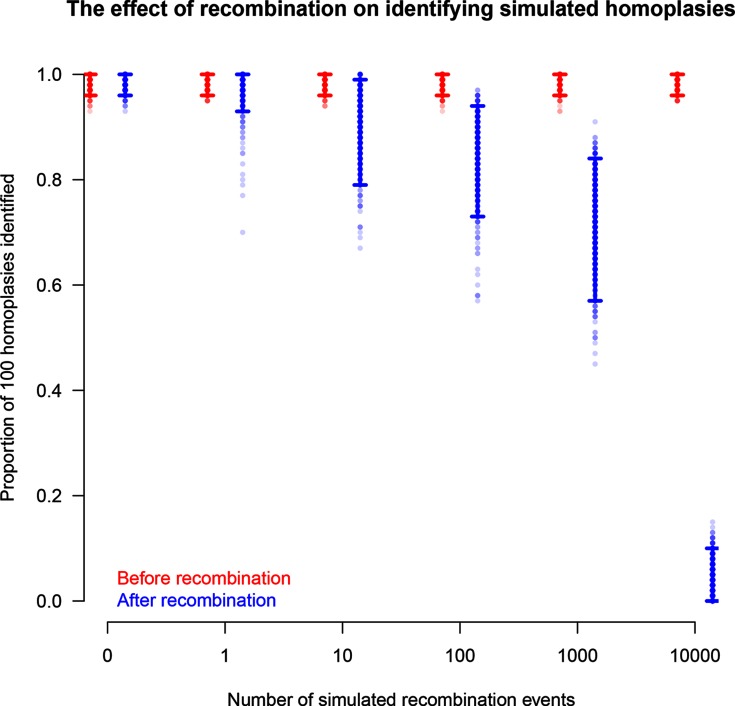
A homoplasy is a nucleotide identity resulting from a process other than inheritance from a common ancestor. Importantly, by distorting the ancestral relationships between nucleotide sequences, homoplasies can change the structure of the phylogeny. Homoplasies can emerge naturally, especially under high selection pressures and/or high mutation rates, or be created during the generation and processing of sequencing data. Identification of homoplasies is critical, both to understand their influence on the analyses of phylogenetic data and to allow an investigation into how they arose. Here we present HomoplasyFinder, a java application that can be used as a stand-a-lone tool or within the statistical programming environment R. Within R and Java, HomoplasyFinder is shown to be able to automatically, and quickly, identify any homoplasies present in simulated and real phylogenetic data. HomoplasyFinder can easily be incorporated into existing analysis pipelines, either within or outside of R, allowing the user to quickly identify homoplasies to inform downstream analyses and interpretation.
Keywords: HomoplasyFinder; Java; R package; convergence; homoplasy; phylogenetic.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
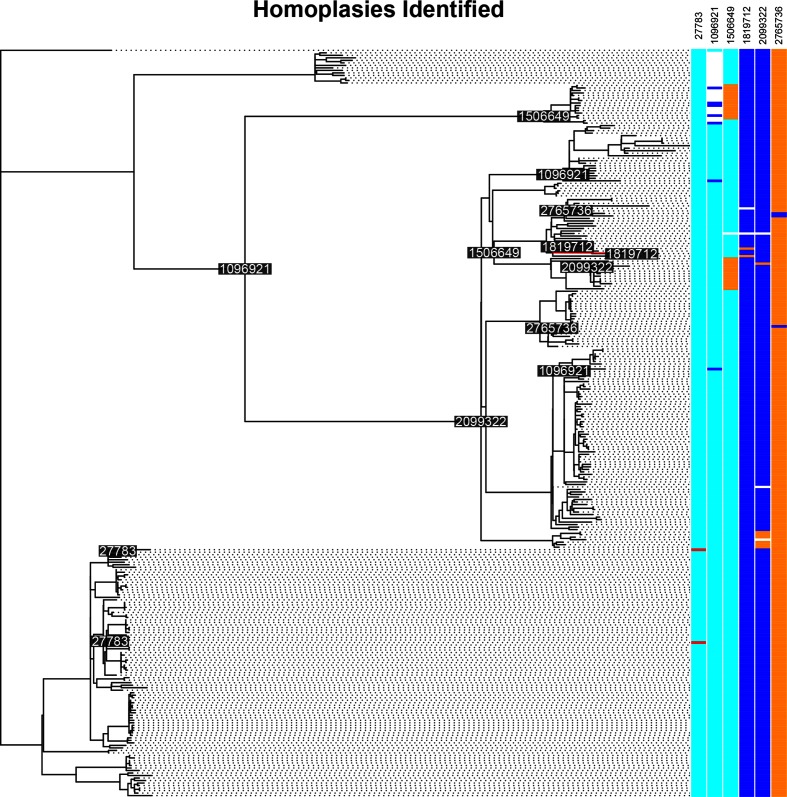
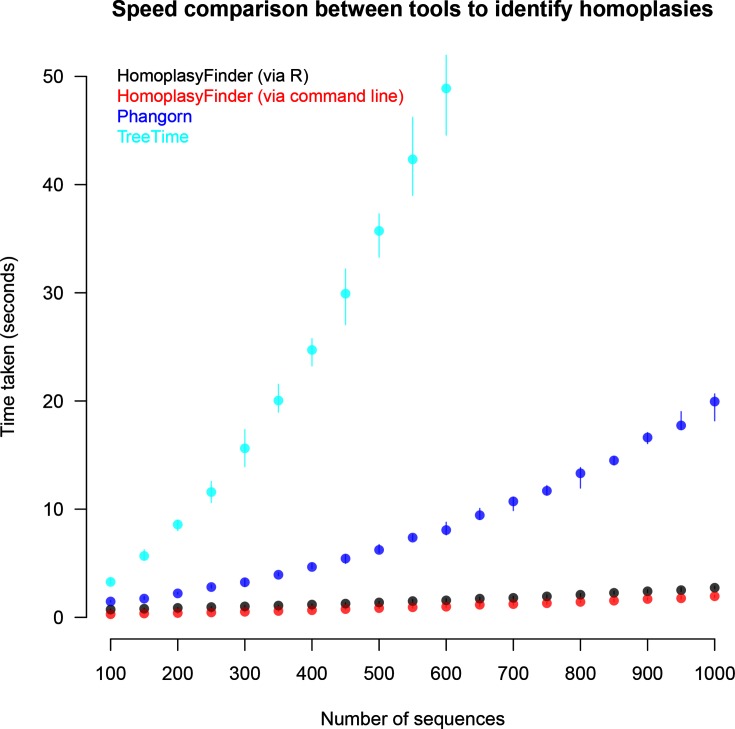
References
-
- Wake DB. Homoplasy: the result of natural selection, or evidence of design limitations? Am Nat. 1991;138:543–567. doi: 10.1086/285234. - DOI
-
- Hassanin A, Lecointre G, Tillier S. The ‘evolutionary signal’ of homoplasy in proteincoding gene sequences and its consequences for a priori weighting in phylogeny. Comptes Rendus de l'Académie des Sciences - Series III - Sciences de la Vie. 1998;321:611–620. doi: 10.1016/S0764-4469(98)80464-2. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources