

Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap
- PMID: 30664679
- PMCID: PMC6607905
- DOI: 10.1038/s41596-018-0103-9
Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap
Abstract
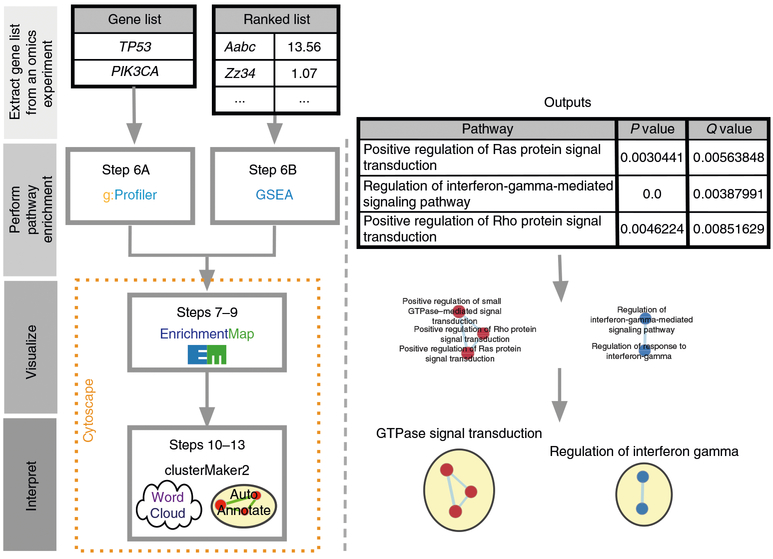
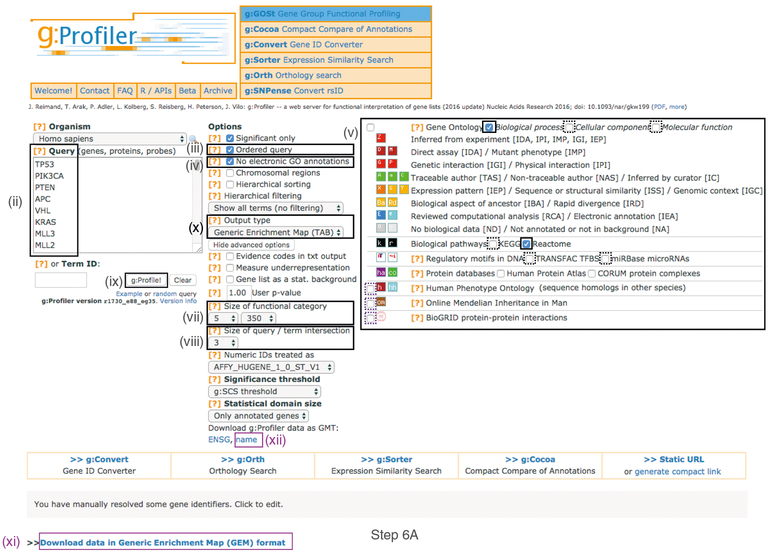
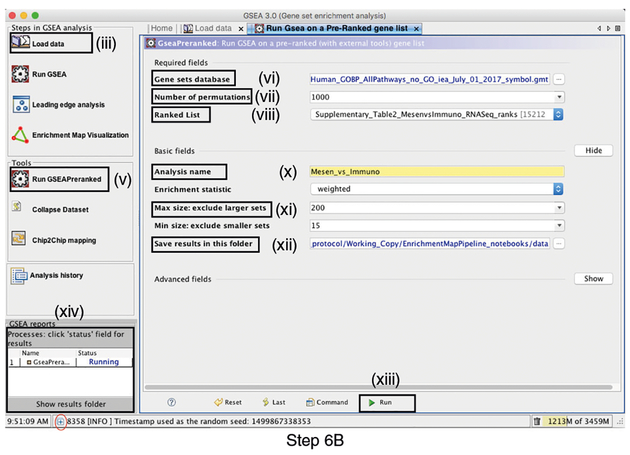
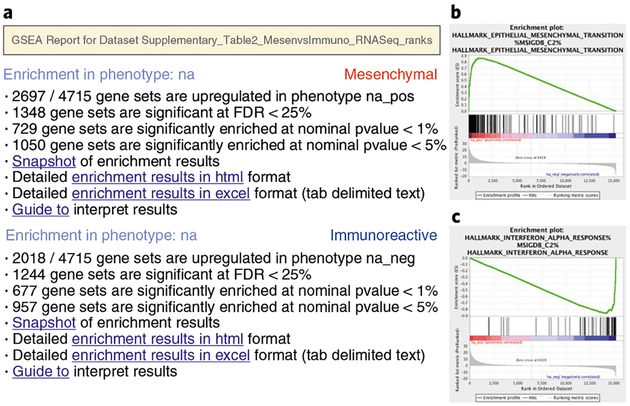
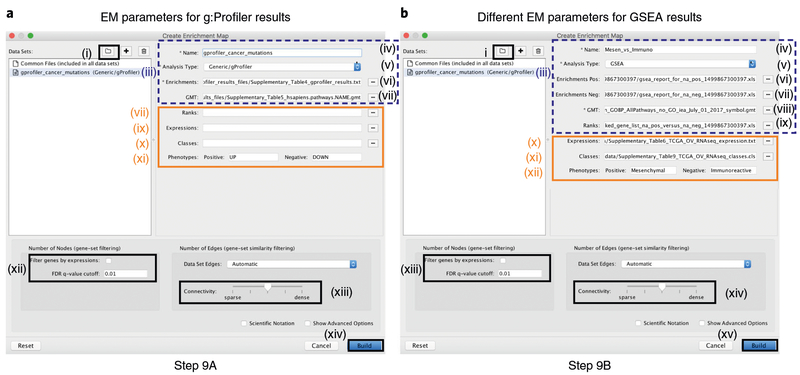
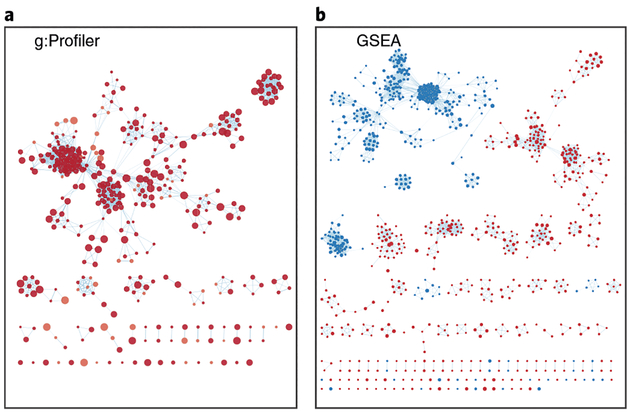
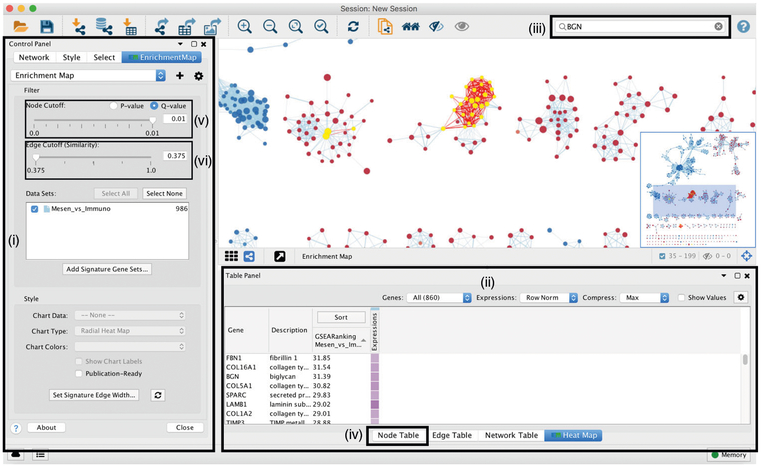
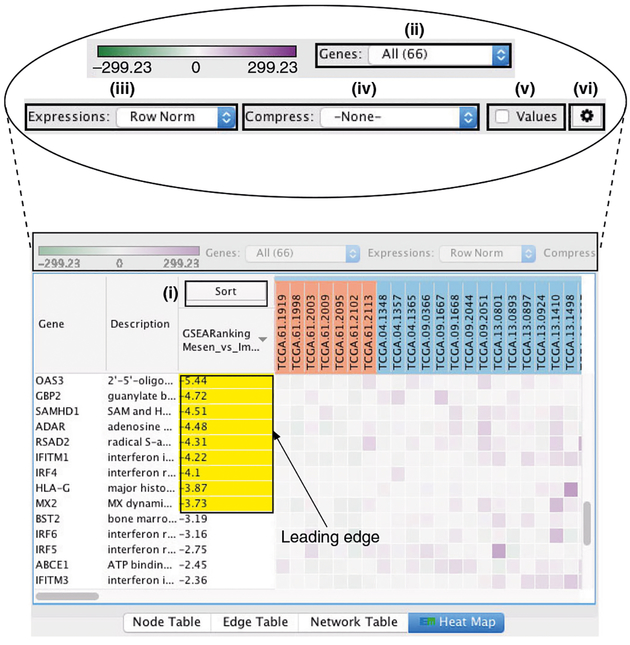
Pathway enrichment analysis helps researchers gain mechanistic insight into gene lists generated from genome-scale (omics) experiments. This method identifies biological pathways that are enriched in a gene list more than would be expected by chance. We explain the procedures of pathway enrichment analysis and present a practical step-by-step guide to help interpret gene lists resulting from RNA-seq and genome-sequencing experiments. The protocol comprises three major steps: definition of a gene list from omics data, determination of statistically enriched pathways, and visualization and interpretation of the results. We describe how to use this protocol with published examples of differentially expressed genes and mutated cancer genes; however, the principles can be applied to diverse types of omics data. The protocol describes innovative visualization techniques, provides comprehensive background and troubleshooting guidelines, and uses freely available and frequently updated software, including g:Profiler, Gene Set Enrichment Analysis (GSEA), Cytoscape and EnrichmentMap. The complete protocol can be performed in ~4.5 h and is designed for use by biologists with no prior bioinformatics training.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures

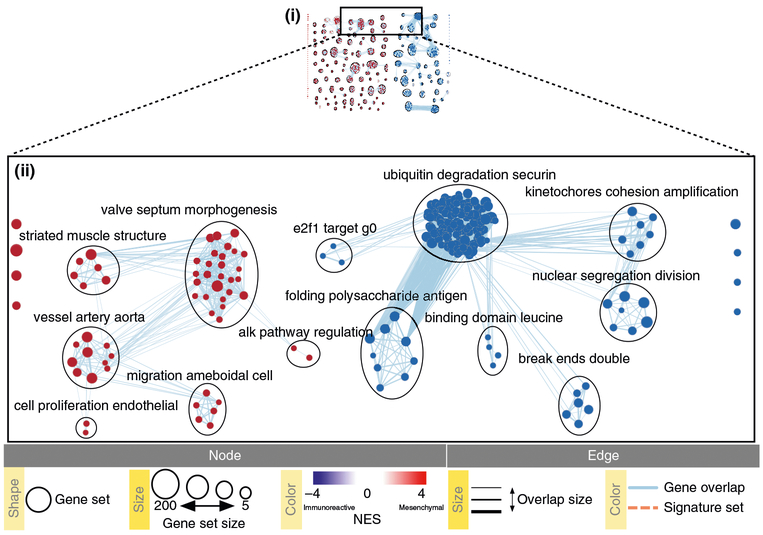
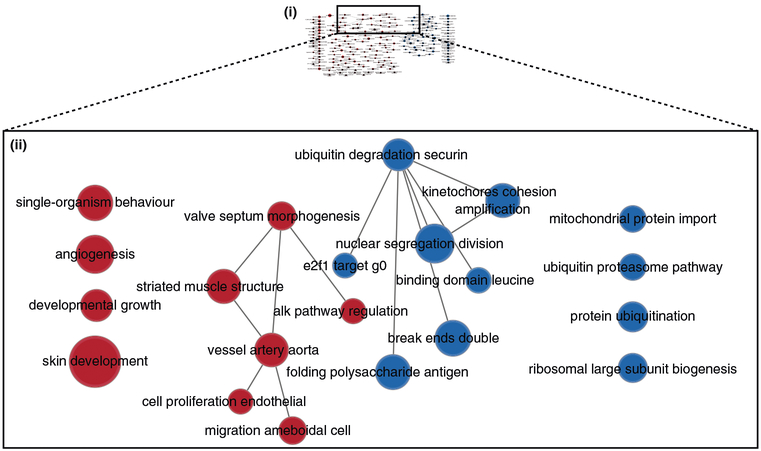
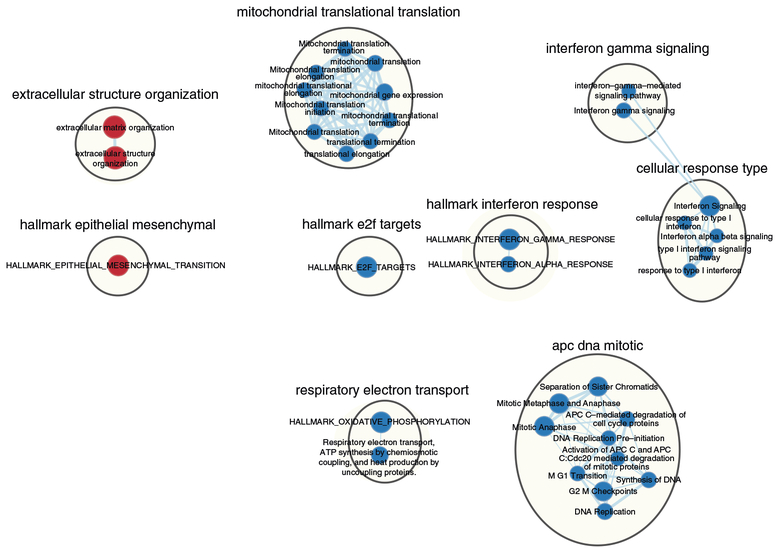
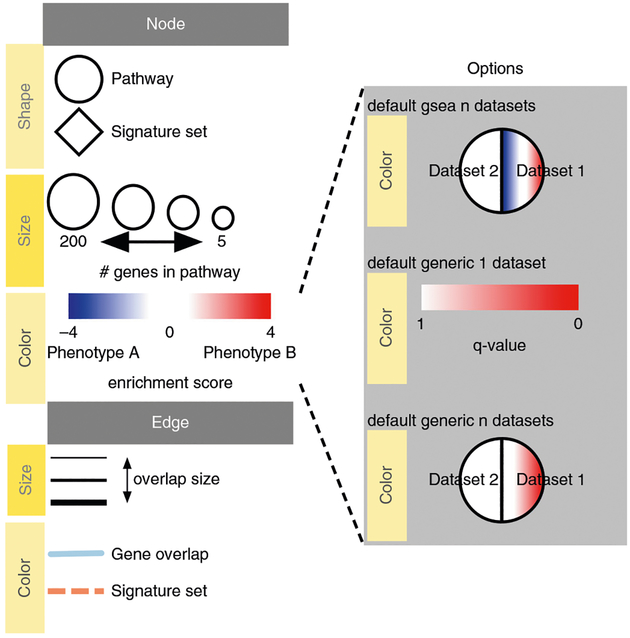
References
-
- Lander ES Initial impact of the sequencing of the human genome. Nature 470, 187–197 (2011). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
