

Commonality despite exceptional diversity in the baseline human antibody repertoire
- PMID: 30664748
- PMCID: PMC6411386
- DOI: 10.1038/s41586-019-0879-y
Commonality despite exceptional diversity in the baseline human antibody repertoire
Abstract
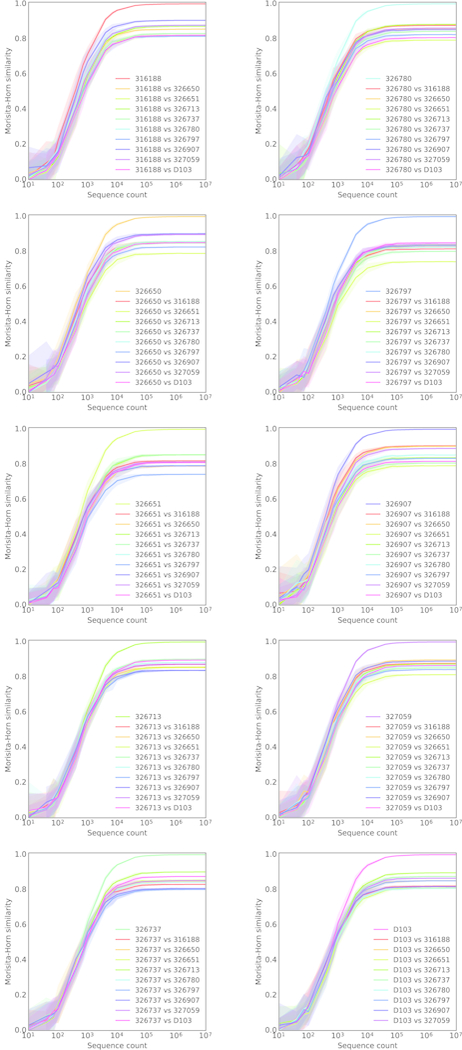
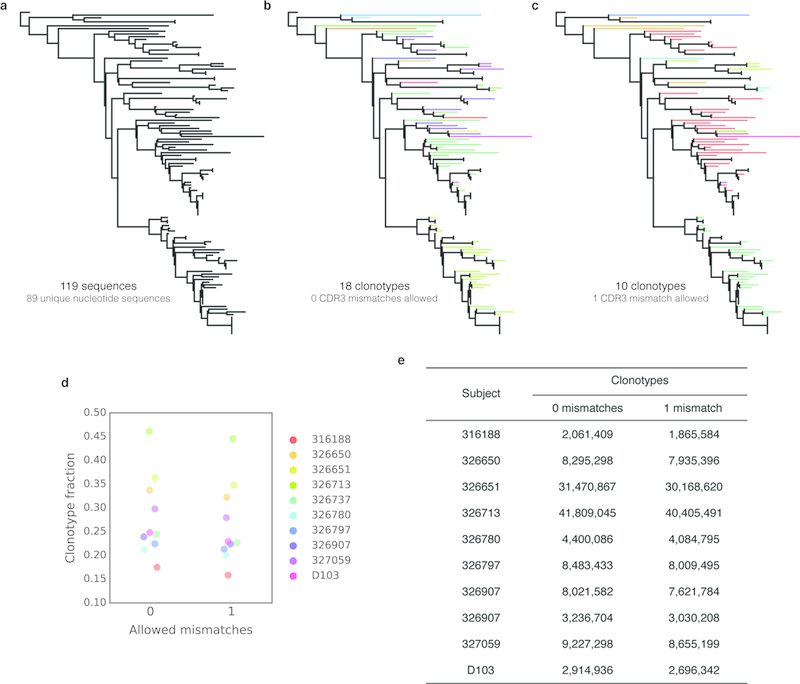
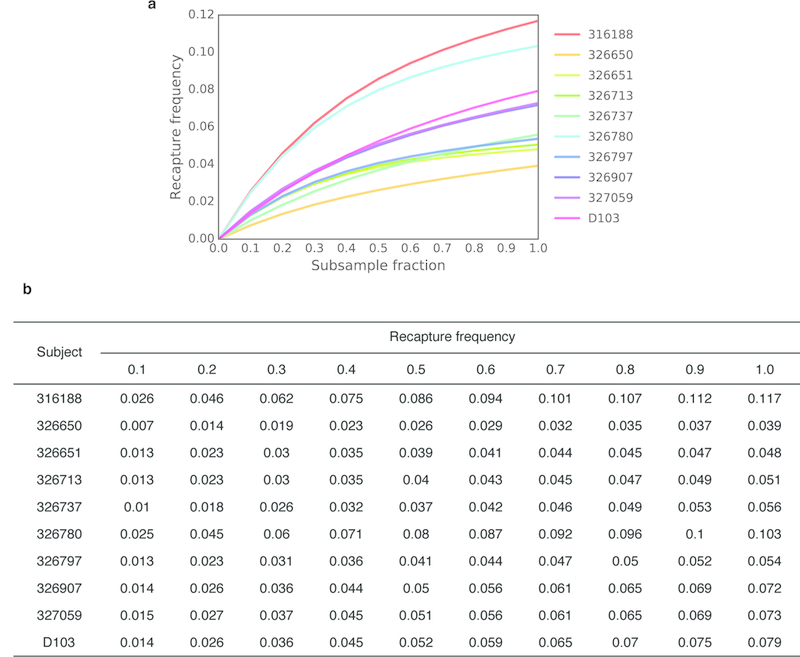
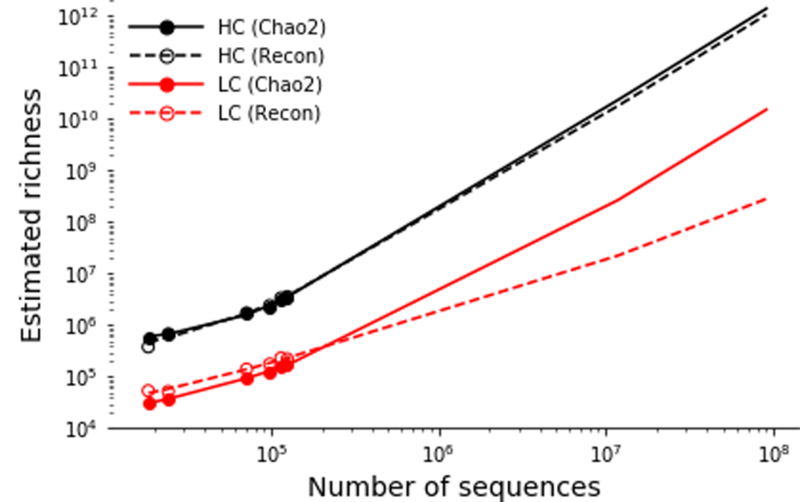
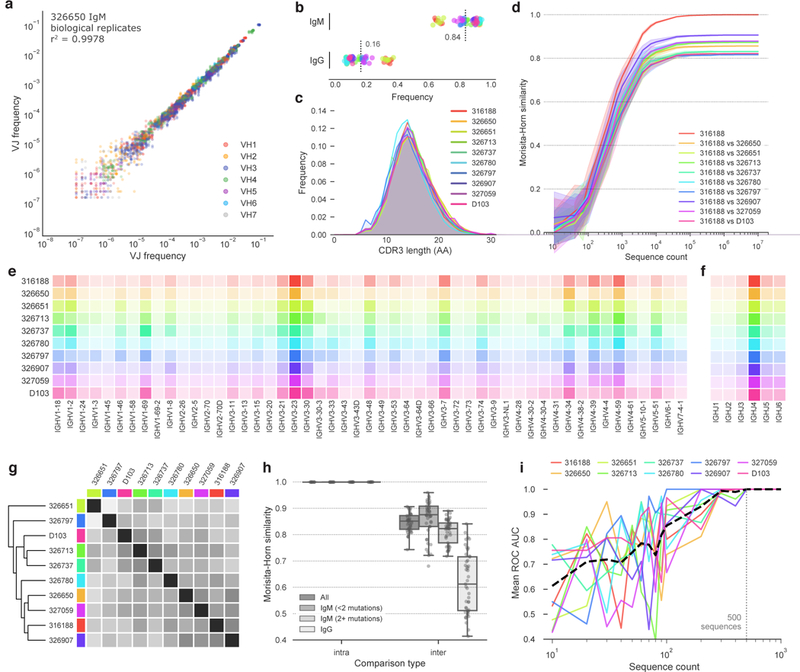
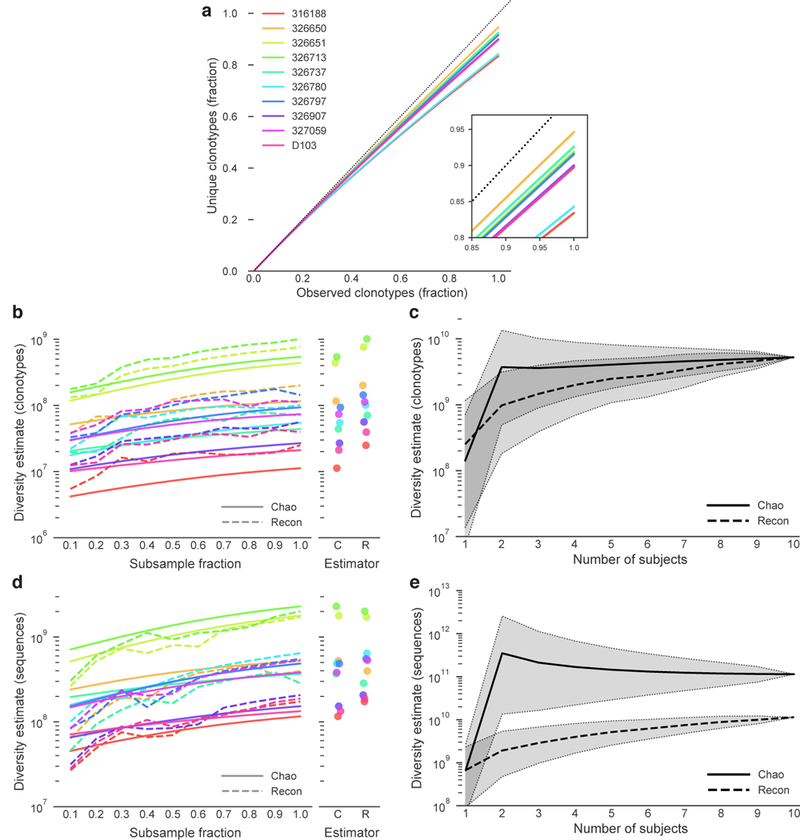
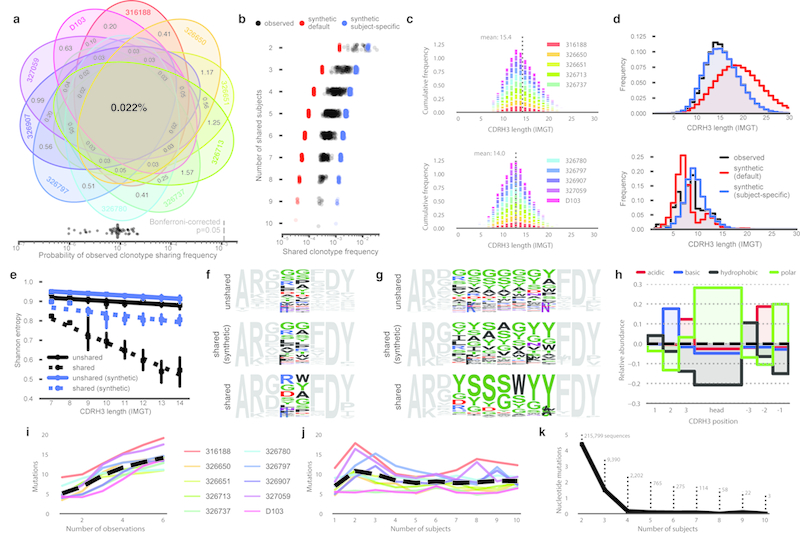
In principle, humans can produce an antibody response to any non-self-antigen molecule in the appropriate context. This flexibility is achieved by the presence of a large repertoire of naive antibodies, the diversity of which is expanded by somatic hypermutation following antigen exposure1. The diversity of the naive antibody repertoire in humans is estimated to be at least 1012 unique antibodies2. Because the number of peripheral blood B cells in a healthy adult human is on the order of 5 × 109, the circulating B cell population samples only a small fraction of this diversity. Full-scale analyses of human antibody repertoires have been prohibitively difficult, primarily owing to their massive size. The amount of information encoded by all of the rearranged antibody and T cell receptor genes in one person-the 'genome' of the adaptive immune system-exceeds the size of the human genome by more than four orders of magnitude. Furthermore, because much of the B lymphocyte population is localized in organs or tissues that cannot be comprehensively sampled from living subjects, human repertoire studies have focused on circulating B cells3. Here we examine the circulating B cell populations of ten human subjects and present what is, to our knowledge, the largest single collection of adaptive immune receptor sequences described to date, comprising almost 3 billion antibody heavy-chain sequences. This dataset enables genetic study of the baseline human antibody repertoire at an unprecedented depth and granularity, which reveals largely unique repertoires for each individual studied, a subpopulation of universally shared antibody clonotypes, and an exceptional overall diversity of the antibody repertoire.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
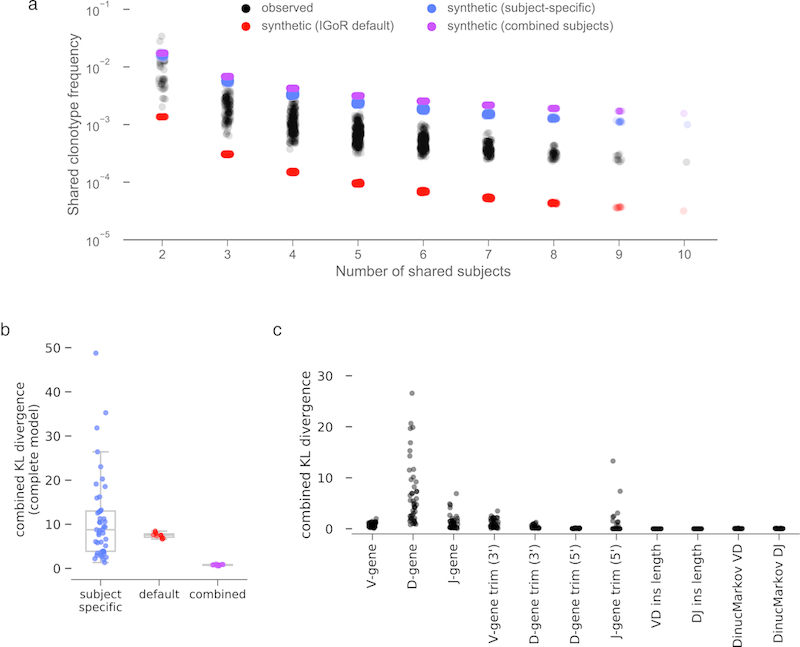
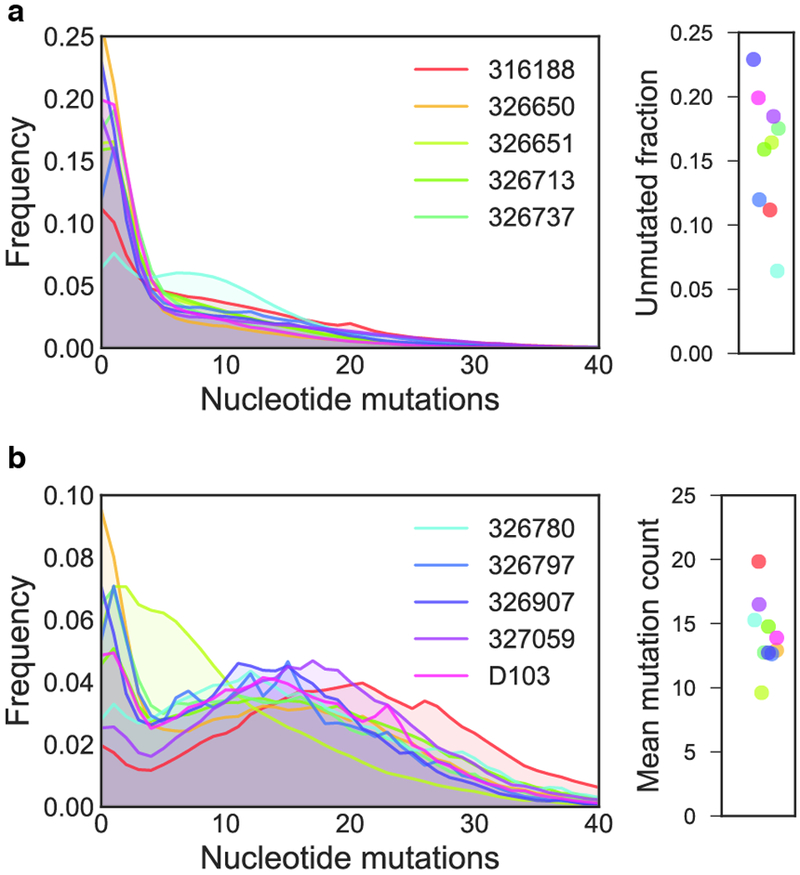
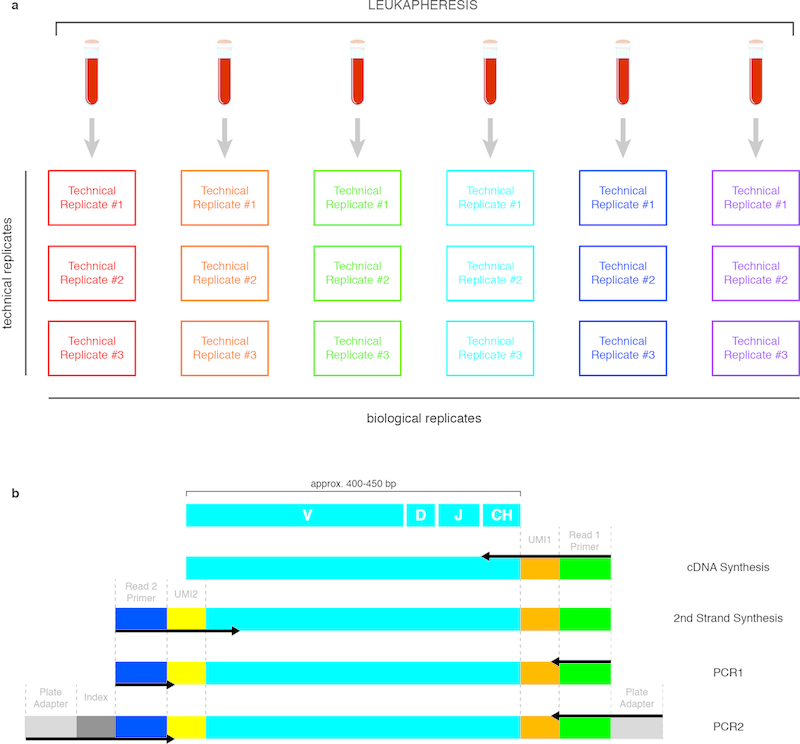
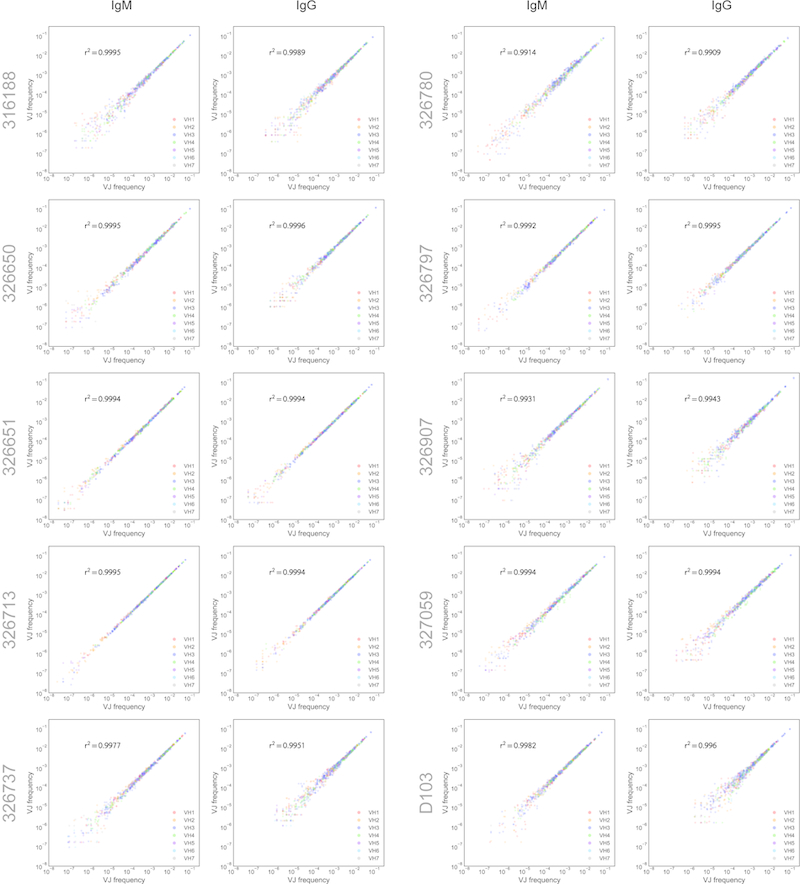
References
-
- Rajewsky K Clonal selection and learning in the antibody system. Nature 381, 751–758 (1996). - PubMed
-
- Alberts B et al. The Generation of Antibody Diversity. (Garland Science, 2002).
-
- Briney B & Burton D Massively scalable genetic analysis of antibody repertoires. bioRxiv 447813 (2018). doi: 10.1101/447813 - DOI
EXTENDED DATA REFERENCES
-
- van Dongen JJM et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98–3936. Leukemia 17, 2257–2317 (2003). - PubMed
-
- Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
-
- DeKosky BJ et al. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat. Med. 21, nm3743 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
