

Selective Ablation of Megalin in the Retinal Pigment Epithelium Results in Megaophthalmos, Macromelanosome Formation and Severe Retina Degeneration
- PMID: 30665232
- PMCID: PMC6343679
- DOI: 10.1167/iovs.18-25667
Selective Ablation of Megalin in the Retinal Pigment Epithelium Results in Megaophthalmos, Macromelanosome Formation and Severe Retina Degeneration
Abstract
Purpose: Mutations in the megalin-encoding gene, LRP2, cause high myopia as seen in patients suffering from Donnai-Barrow/facio-oculo-acoustico-renal syndrome. Megalin is present in both the nonpigmented epithelium of the ciliary body and in the RPE. In this study, we set out to establish an animal model to study the mechanisms underlying the ocular phenotype and to establish if high myopia/megaophthalmos is induced by postnatal megalin-deficiency in the RPE.
Methods: Postnatal RPE-specific deletion of megalin was generated by crossing mice bearing a homozygous loxP-flanked Lrp2 allele with transgenic mice expressing the Cre recombinase driven by the BEST1 promotor. The model was investigated by immunohistologic techniques, and transmission electron microscopy.
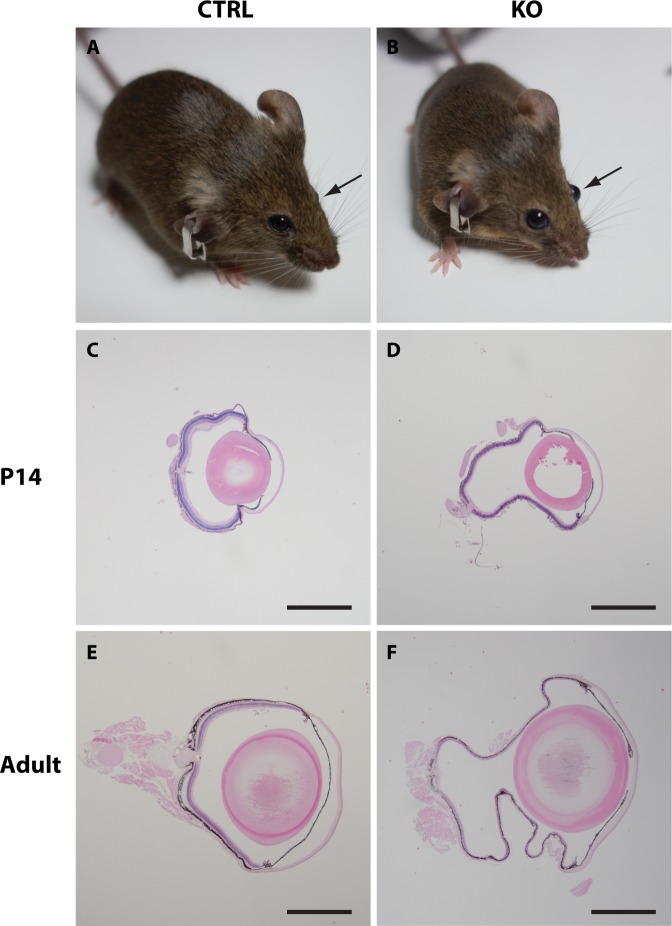
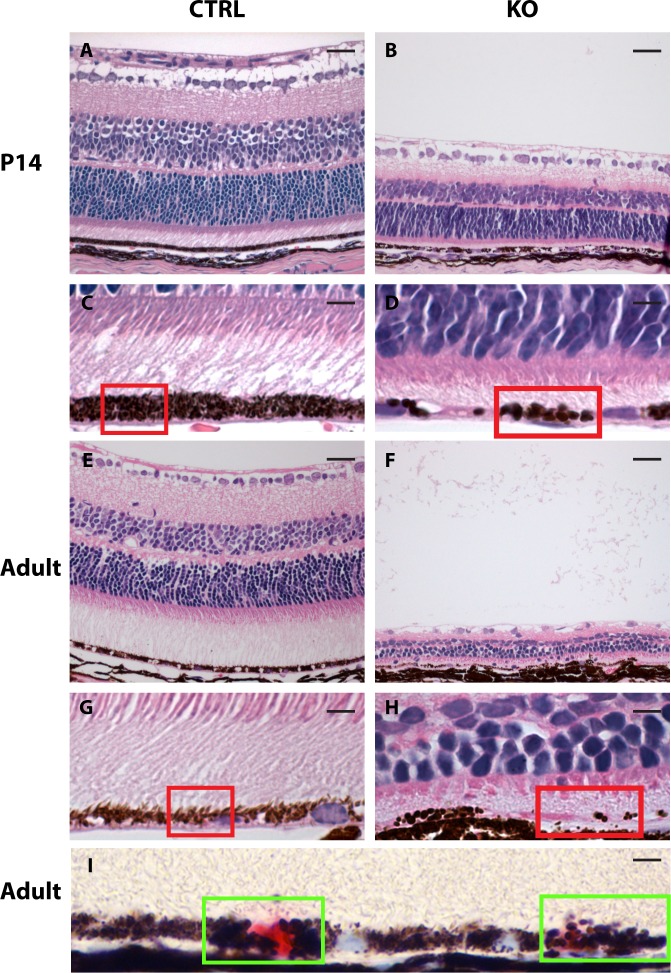
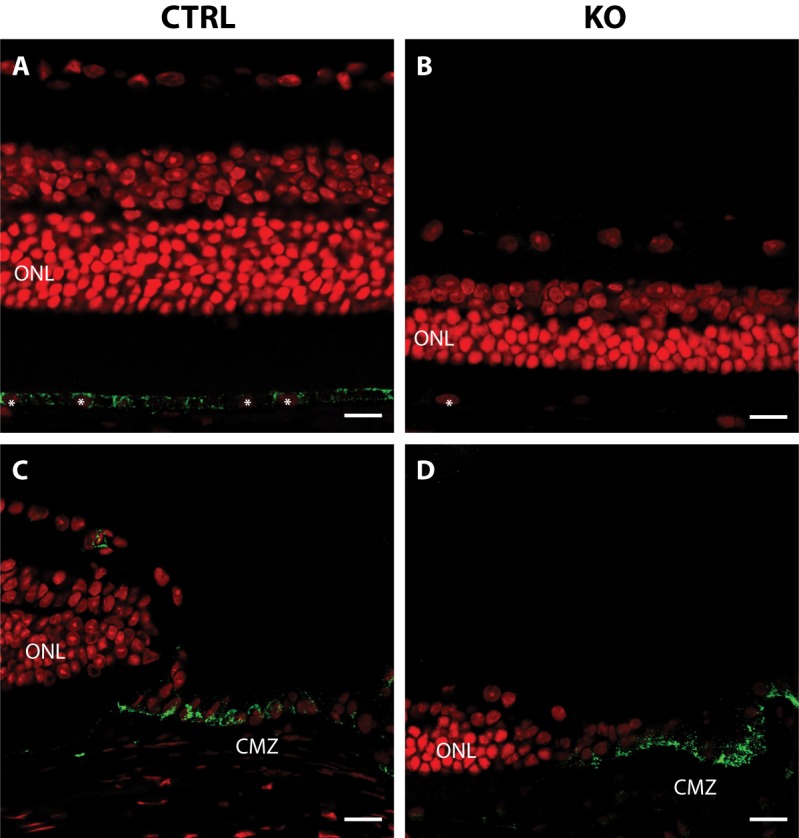
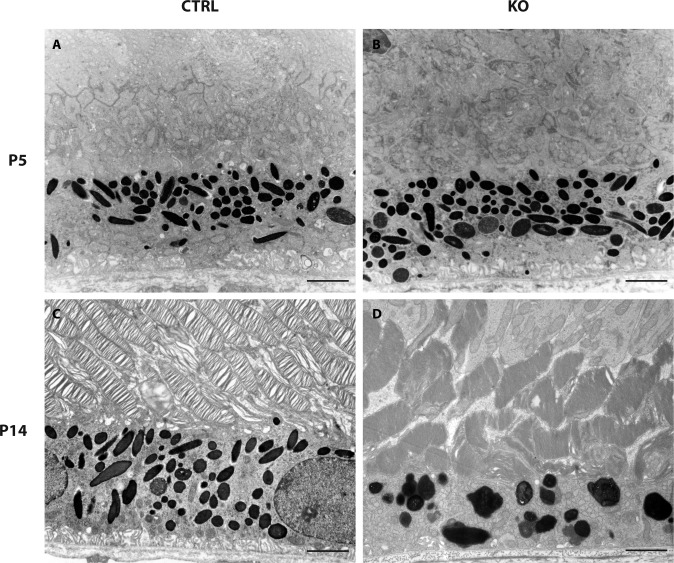
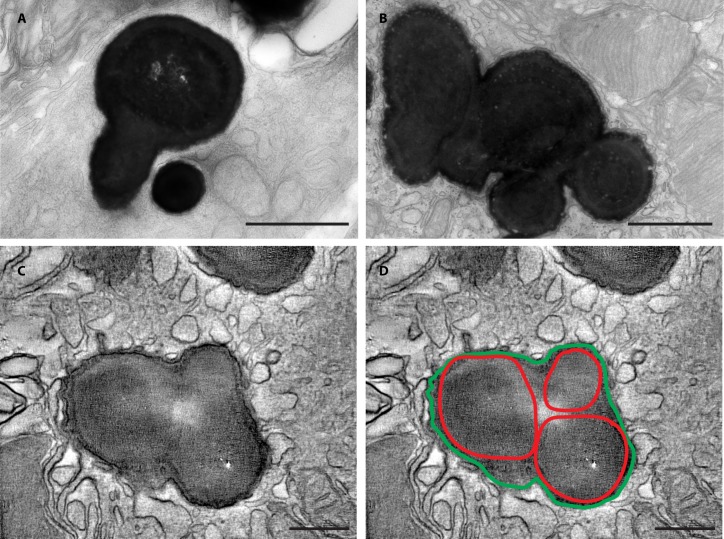
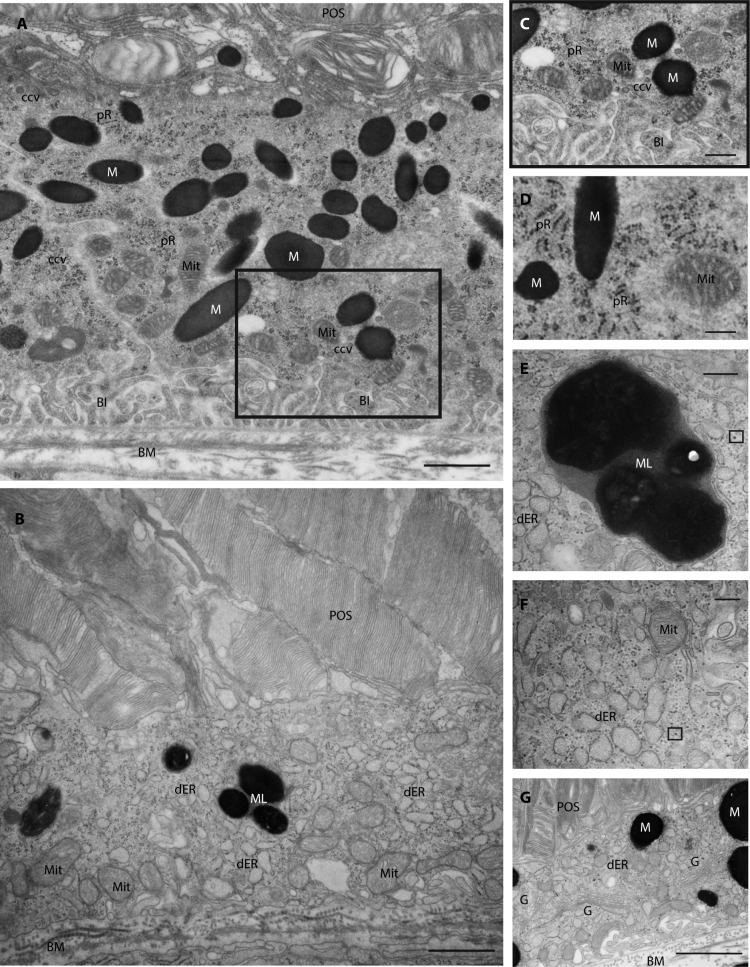
Results: Mice with postnatal RPE-specific loss of megalin developed a megaophthalmos phenotype with dramatic increase in ocular size and severe retinal thinning associated with compromised vision. This phenotype was present at postnatal day 14, indicating rapid development in the period from onset of BEST1 promotor activity at postnatal day 10. Additionally, RPE melanosomes exhibited abnormal size and morphology, suggested by electron tomography to be caused by fusion events between multiple melanosomes.
Conclusions: Postnatal loss of megalin in the RPE induces dramatic and rapid ocular growth and retinal degeneration compatible with the high myopia observed in Donnai-Barrow patients. The morphologic changes of RPE melanosomes, believed to be largely inert and fully differentiated at birth, suggested a continued plasticity of mature melanosomes and a requirement for megalin to maintain their number and morphology.
Figures
References
-
- Patel N, Hejkal T, Katz A, Margalit E. Ocular manifestations of Donnai-Barrow syndrome. J Child Neurol. 2007;22:462–464. - PubMed
-
- Schrauwen I, Sommen M, Claes C, et al. Broadening the phenotype of LRP2 mutations: a new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clin Genet. 2013;86:282–286. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
