

Integrating predicted transcriptome from multiple tissues improves association detection
- PMID: 30668570
- PMCID: PMC6358100
- DOI: 10.1371/journal.pgen.1007889
Integrating predicted transcriptome from multiple tissues improves association detection
Abstract
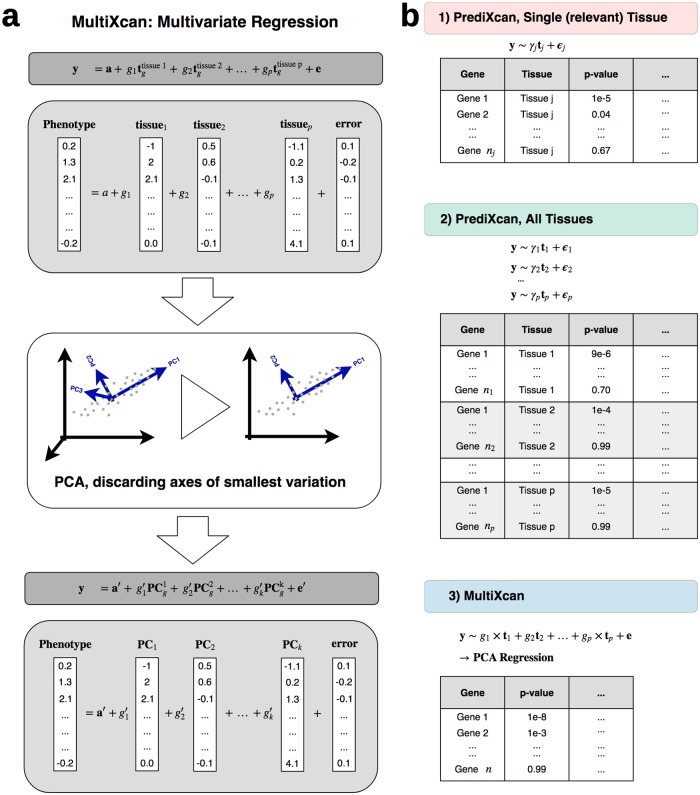
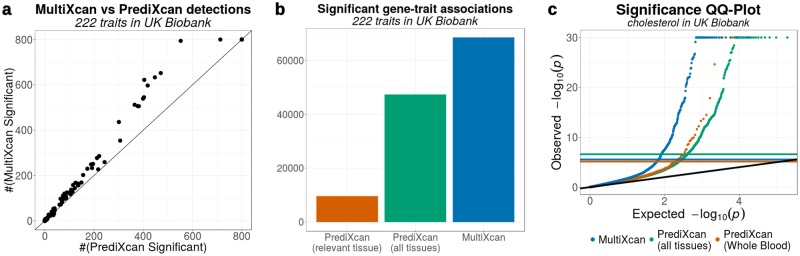
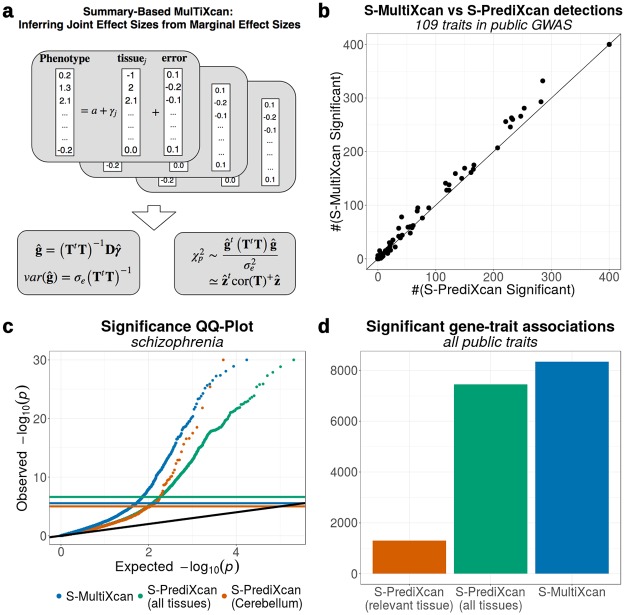
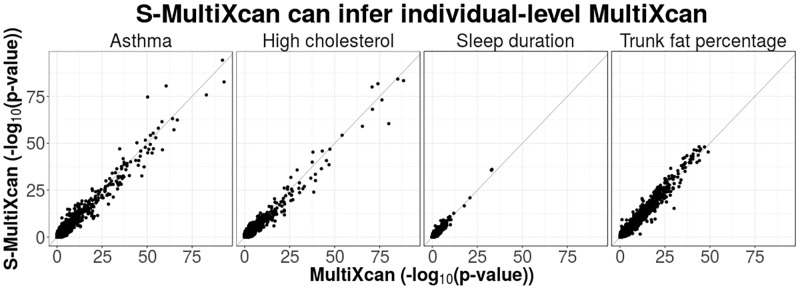
Integration of genome-wide association studies (GWAS) and expression quantitative trait loci (eQTL) studies is needed to improve our understanding of the biological mechanisms underlying GWAS hits, and our ability to identify therapeutic targets. Gene-level association methods such as PrediXcan can prioritize candidate targets. However, limited eQTL sample sizes and absence of relevant developmental and disease context restrict our ability to detect associations. Here we propose an efficient statistical method (MultiXcan) that leverages the substantial sharing of eQTLs across tissues and contexts to improve our ability to identify potential target genes. MultiXcan integrates evidence across multiple panels using multivariate regression, which naturally takes into account the correlation structure. We apply our method to simulated and real traits from the UK Biobank and show that, in realistic settings, we can detect a larger set of significantly associated genes than using each panel separately. To improve applicability, we developed a summary result-based extension called S-MultiXcan, which we show yields highly concordant results with the individual level version when LD is well matched. Our multivariate model-based approach allowed us to use the individual level results as a gold standard to calibrate and develop a robust implementation of the summary-based extension. Results from our analysis as well as software and necessary resources to apply our method are publicly available.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Nurnberger JI, et al. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371–9. Available from: 026E30F$nhttp://www.ncbi.nlm.nih.gov/pubmed/23453885">http://discovery.u.... - PMC - PubMed
-
- Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nature genetics. 2013;45(1):25–33. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3679547{&}tool.... - PMC - PubMed
-
- Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nature Genetics. 2012;44(9):981–990. Available from: 026E30F$nhttp://www.nature.com/doifinder/10.1038/ng.2383">http://www.ncb.... - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
