

Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer's disease
- PMID: 30670054
- PMCID: PMC6343260
- DOI: 10.1186/s13024-019-0307-7
Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer's disease
Abstract
Background: Identifying effective strategies to prevent memory loss in AD has eluded researchers to date, and likely reflects insufficient understanding of early pathogenic mechanisms directly affecting memory encoding. As synaptic loss best correlates with memory loss in AD, refocusing efforts to identify factors driving synaptic impairments may provide the critical insight needed to advance the field. In this study, we reveal a previously undescribed cascade of events underlying pre and postsynaptic hippocampal signaling deficits linked to cognitive decline in AD. These profound alterations in synaptic plasticity, intracellular Ca2+ signaling, and network propagation are observed in 3-4 month old 3xTg-AD mice, an age which does not yet show overt histopathology or major behavioral deficits.
Methods: In this study, we examined hippocampal synaptic structure and function from the ultrastructural level to the network level using a range of techniques including electron microscopy (EM), patch clamp and field potential electrophysiology, synaptic immunolabeling, spine morphology analyses, 2-photon Ca2+ imaging, and voltage-sensitive dye-based imaging of hippocampal network function in 3-4 month old 3xTg-AD and age/background strain control mice.
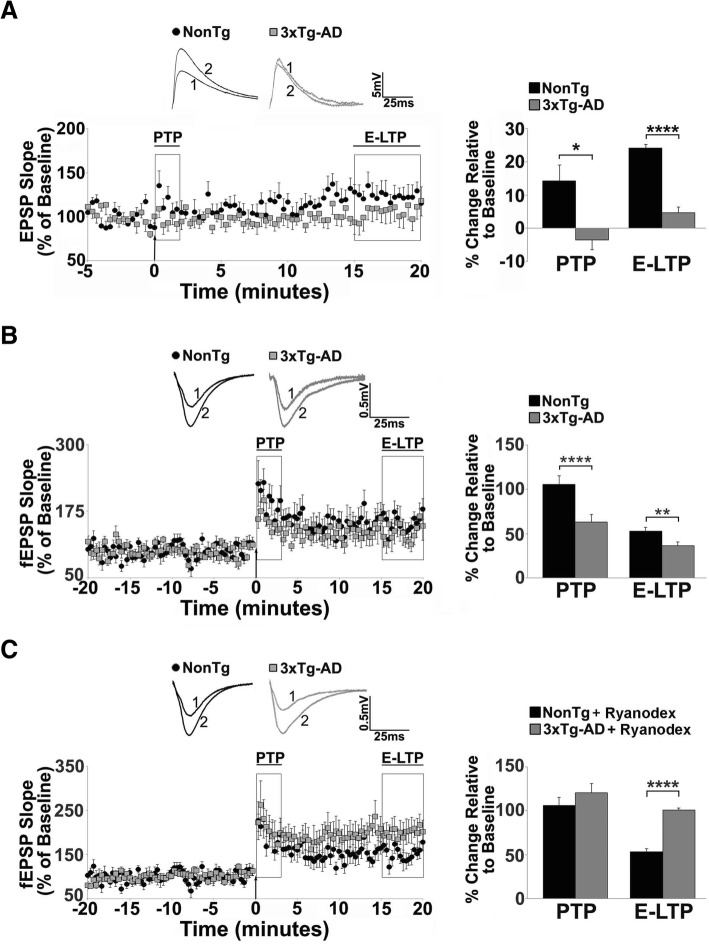
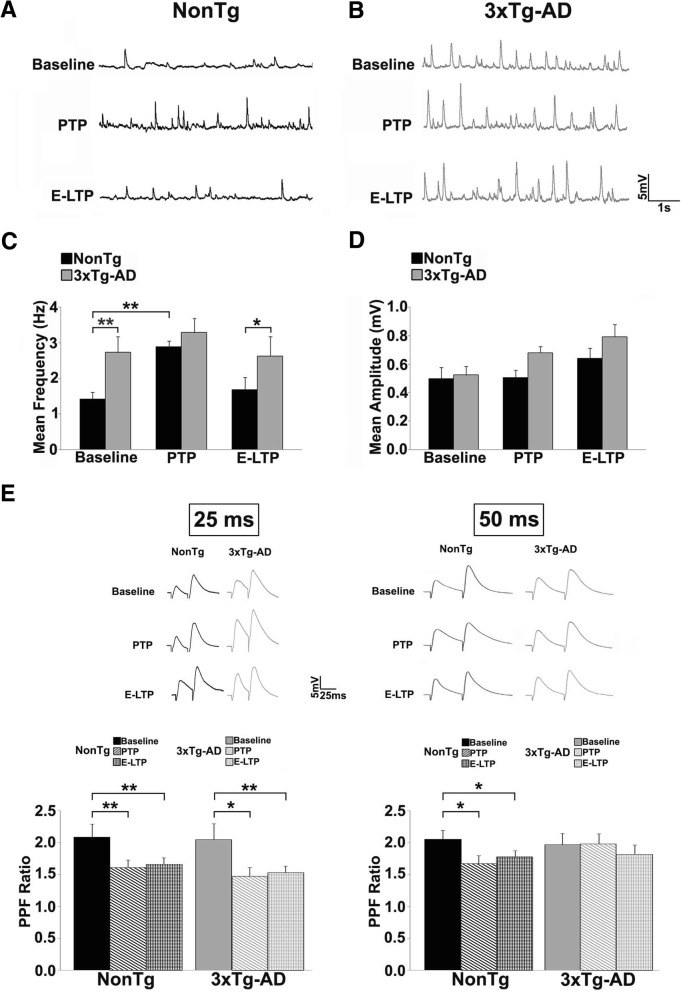
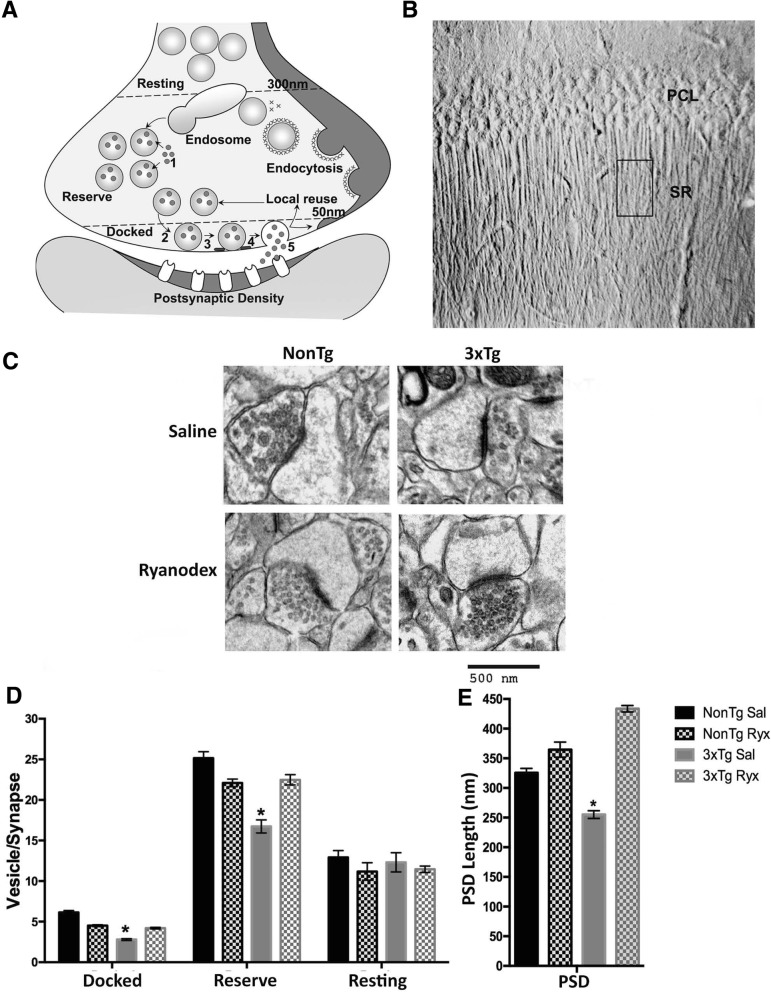
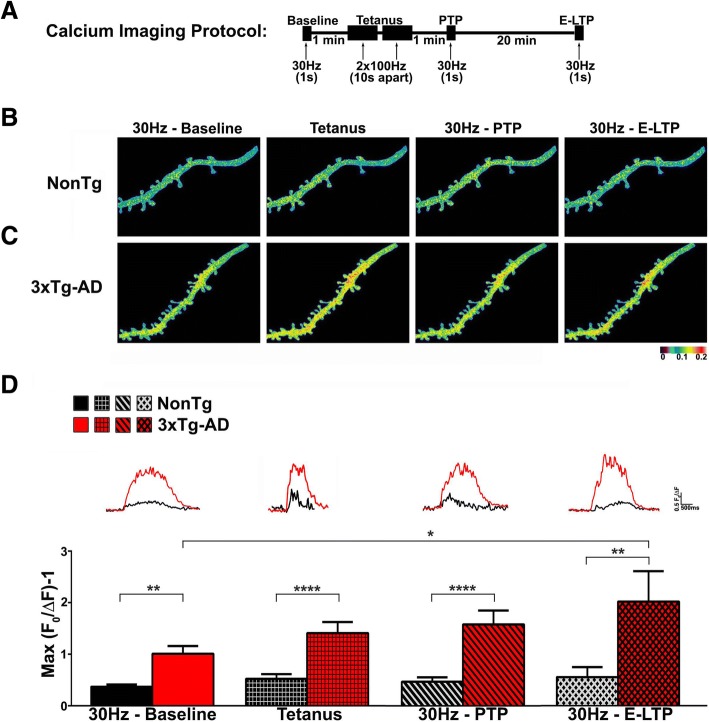
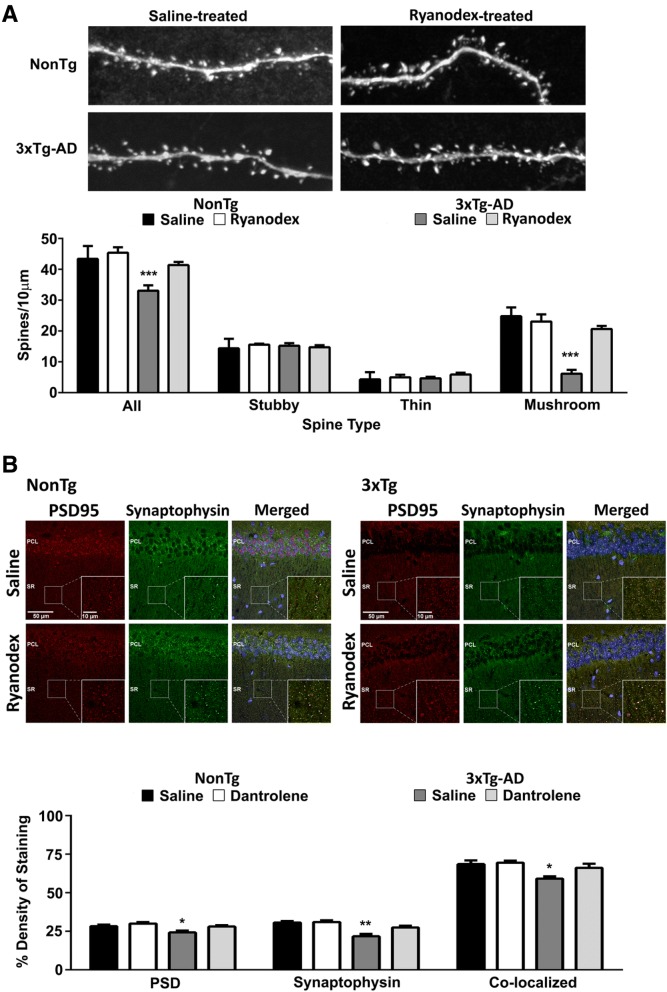
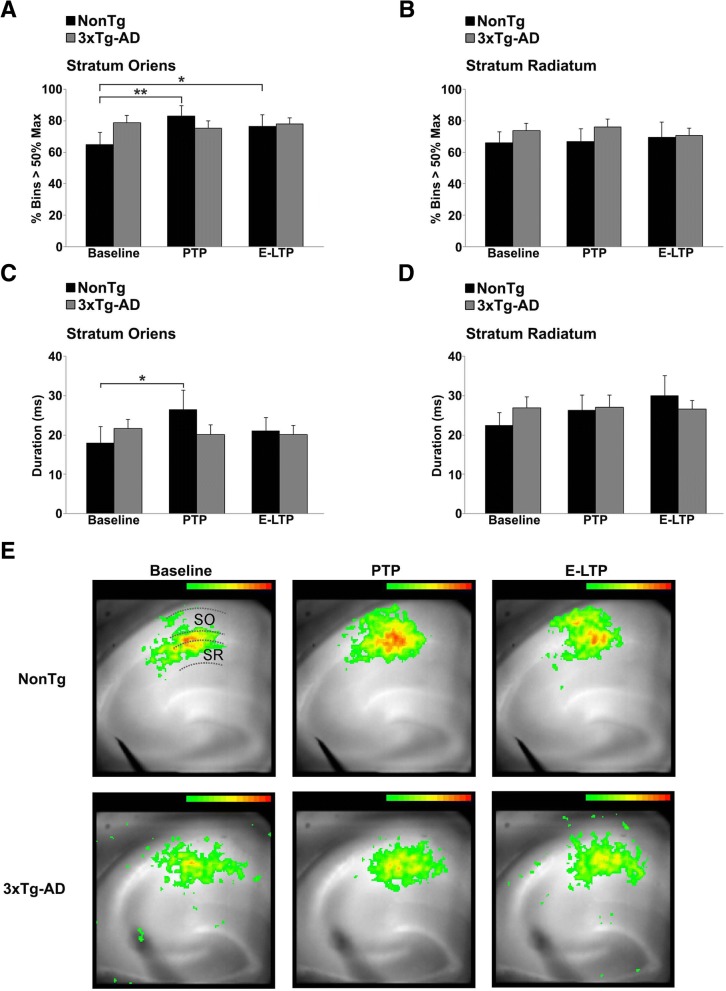
Results: In 3xTg-AD mice, short-term plasticity at the CA1-CA3 Schaffer collateral synapse is profoundly impaired; this has broader implications for setting long-term plasticity thresholds. Alterations in spontaneous vesicle release and paired-pulse facilitation implicated presynaptic signaling abnormalities, and EM analysis revealed a reduction in the ready-releasable and reserve pools of presynaptic vesicles in CA3 terminals; this is an entirely new finding in the field. Concurrently, increased synaptically-evoked Ca2+ in CA1 spines triggered by LTP-inducing tetani is further enhanced during PTP and E-LTP epochs, and is accompanied by impaired synaptic structure and spine morphology. Notably, vesicle stores, synaptic structure and short-term plasticity are restored by normalizing intracellular Ca2+ signaling in the AD mice.
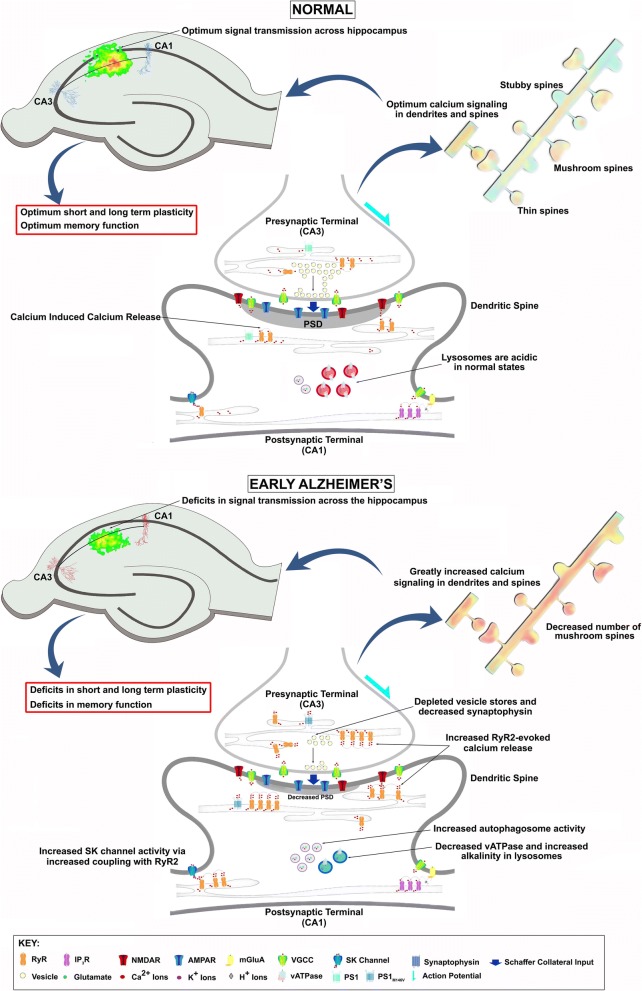
Conclusions: These findings suggest the Ca2+ dyshomeostasis within synaptic compartments has an early and fundamental role in driving synaptic pathophysiology in early stages of AD, and may thus reflect a foundational disease feature driving later cognitive impairment. The overall significance is the identification of previously unidentified defects in pre and postsynaptic compartments affecting synaptic vesicle stores, synaptic plasticity, and network propagation, which directly impact memory encoding.
Keywords: 2-photon imaging; Alzheimer’s disease; Calcium; Electron microscopy; Hippocampus; Mouse model; Network imaging; Patch clamp; Ryanodine receptor; Short-term plasticity; Spines; Synaptic; Synaptic vesicles.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
