

Genetics of MDS
- PMID: 30670442
- PMCID: PMC6587668
- DOI: 10.1182/blood-2018-10-844621
Genetics of MDS
Abstract
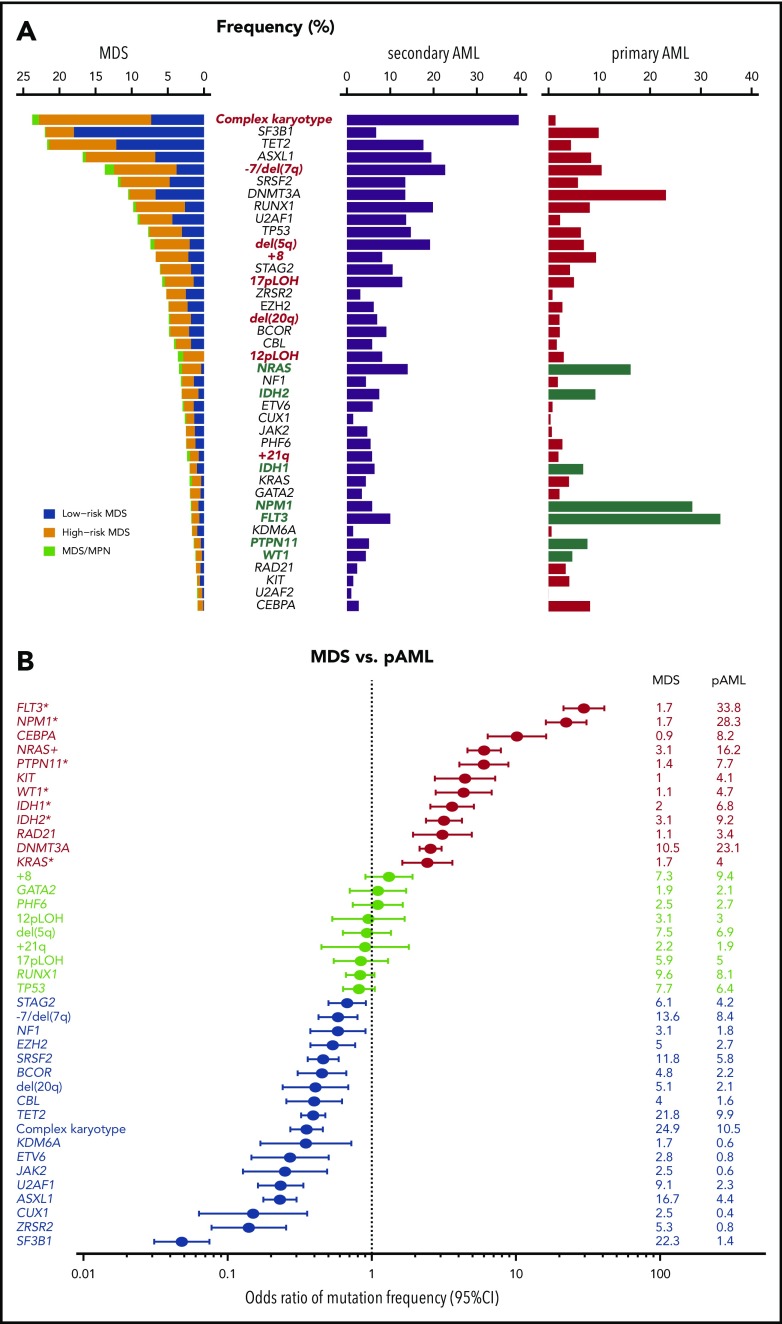
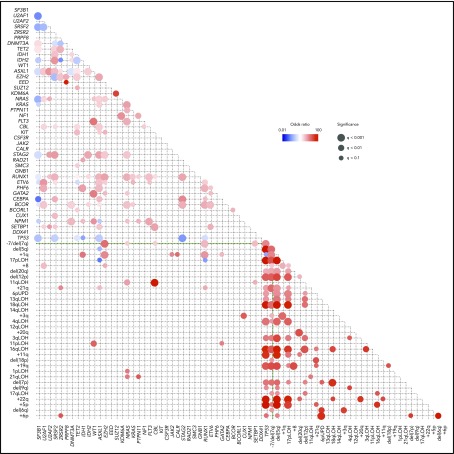
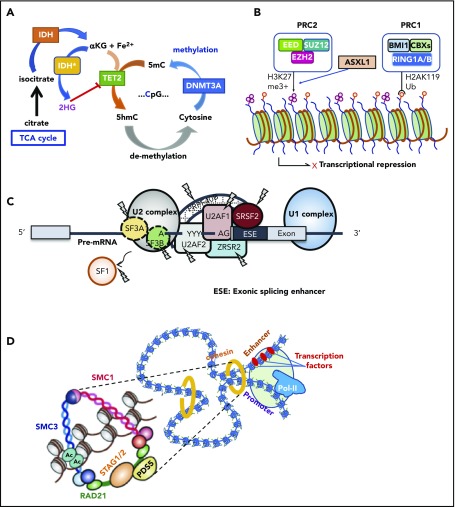
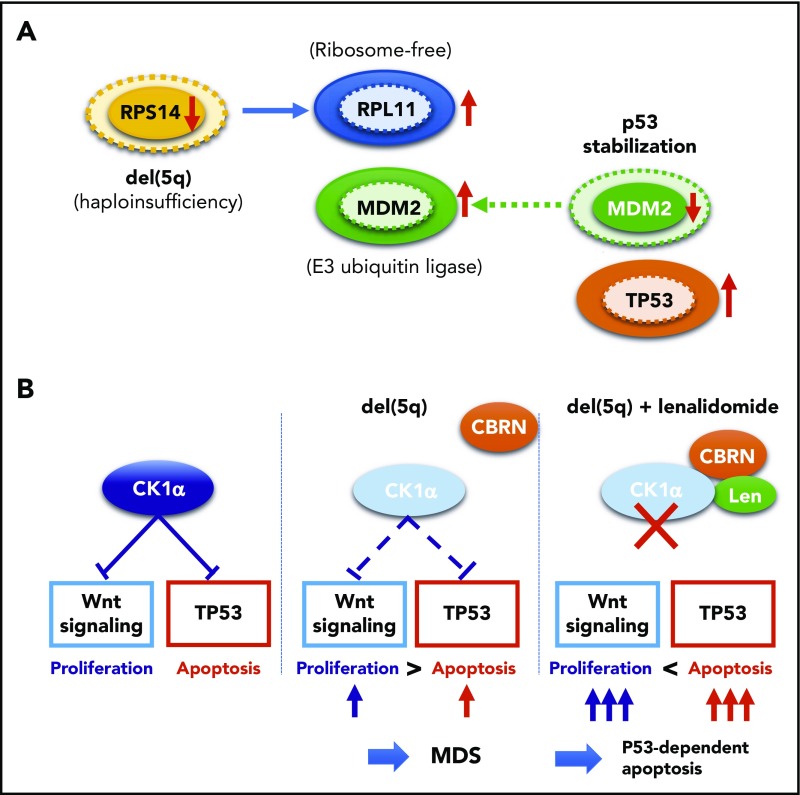
Our knowledge about the genetics of myelodysplastic syndromes (MDS) and related myeloid disorders has been dramatically improved during the past decade, in which revolutionized sequencing technologies have played a major role. Through intensive efforts of sequencing of a large number of MDS genomes, a comprehensive registry of driver mutations recurrently found in a recognizable fraction of MDS patients has been revealed, and ongoing efforts are being made to clarify their impacts on clinical phenotype and prognosis, as well as their role in the pathogenesis of MDS. Among major mutational targets in MDS are the molecules involved in DNA methylations, chromatin modification, RNA splicing, transcription, signal transduction, cohesin regulation, and DNA repair. Showing substantial overlaps with driver mutations seen in acute myeloid leukemia (AML), as well as age-related clonal hematopoiesis in healthy individuals, these mutations are presumed to have a common clonal origin. Mutations are thought to be acquired and positively selected in a well-organized manner to allow for expansion of the initiating clone to compromise normal hematopoiesis, ultimately giving rise to MDS and subsequent transformation to AML in many patients. Significant correlations between mutations suggest the presence of functional interactions between mutations, which dictate disease progression. Mutations are frequently associated with specific disease phenotype, drug response, and clinical outcomes, and thus, it is essential to be familiar with MDS genetics for better management of patients. This review aims to provide a brief overview of the recent progresses in MDS genetics.
© 2019 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The author declares no competing financial interests.
Figures
References
-
- Nimer SD. Myelodysplastic syndromes. Blood. 2008;111(10):4841-4851. - PubMed
-
- Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361(19):1872-1885. - PubMed
-
- McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613-628. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
