

Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases
- PMID: 30670468
- PMCID: PMC6671943
- DOI: 10.1101/cshperspect.a033928
Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases
Abstract
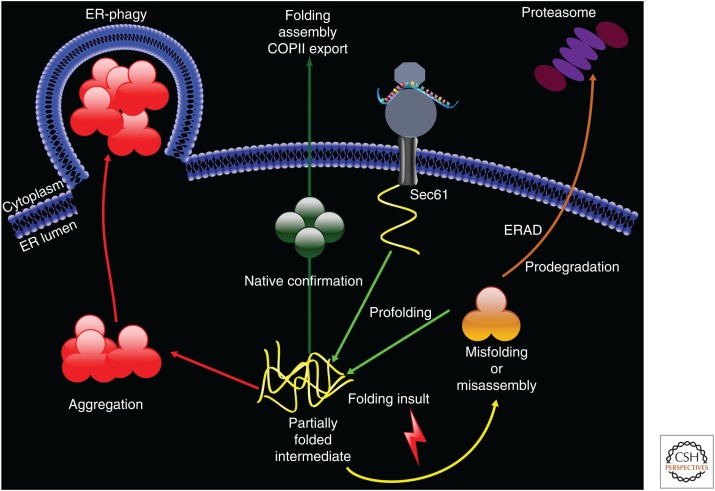
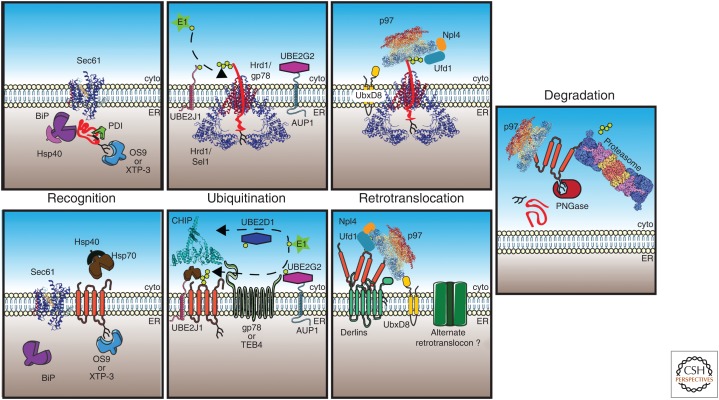
Misfolded proteins compromise cellular homeostasis. This is especially problematic in the endoplasmic reticulum (ER), which is a high-capacity protein-folding compartment and whose function requires stringent protein quality-control systems. Multiprotein complexes in the ER are able to identify, remove, ubiquitinate, and deliver misfolded proteins to the 26S proteasome for degradation in the cytosol, and these events are collectively termed ER-associated degradation, or ERAD. Several steps in the ERAD pathway are facilitated by molecular chaperone networks, and the importance of ERAD is highlighted by the fact that this pathway is linked to numerous protein conformational diseases. In this review, we discuss the factors that constitute the ERAD machinery and detail how each step in the pathway occurs. We then highlight the underlying pathophysiology of protein conformational diseases associated with ERAD.
Copyright © 2019 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Accili D, Kadowaki T, Kadowaki H, Mosthaf L, Ullrich A, Taylor SI. 1992. Immunoglobulin heavy chain-binding protein binds to misfolded mutant insulin receptors with mutations in the extracellular domain. J Biol Chem 267: 586–590. - PubMed
-
- Allen S, Abuzenadah AM, Hinks J, Blagg JL, Gursel T, Ingerslev J, Goodeve AC, Peake IR, Daly ME. 2000. A novel von Willebrand disease–causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood 96: 560–568. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources