

Discovery and Characterization of Mycobacterium basiliense sp. nov., a Nontuberculous Mycobacterium Isolated From Human Lungs
- PMID: 30671031
- PMCID: PMC6331445
- DOI: 10.3389/fmicb.2018.03184
Discovery and Characterization of Mycobacterium basiliense sp. nov., a Nontuberculous Mycobacterium Isolated From Human Lungs
Abstract
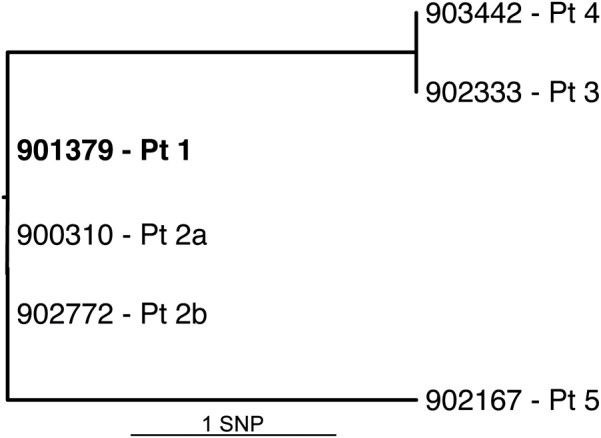
Bacteria belonging to the genus Mycobacterium are predominantly responsible for pulmonary diseases; most notably Mycobacterium tuberculosis causes granulomatous pulmonary infections. Here we describe a novel slow growing mycobacterial species isolated from respiratory samples from five patients, four with underlying pulmonary disease. The isolates were characterized by biochemical and molecular techniques, including whole genome sequencing. Biochemical characteristics generally match those of M. marinum and M. ulcerans; however, the most striking difference of the new species is its ability to grow at 37°C. The new species was found to grow in human macrophages, but not amoebae, suggesting a pathogenic rather than an environmental lifestyle. Phylogenetic analysis reveals a deep-rooting relationship to M. marinum and M. ulcerans. A complete genome sequence was obtained through combining short and long-read sequencing, providing a genome of 5.6 Mb. The genome appears to be highly intact, syntenic with that of M. marinum, with very few insertion sequences. A vast array of virulence factors includes 283 PE/PPE surface-associated proteins, making up 10% of the coding capacity, and 22 non-ribosomal peptide synthase clusters. A comparison of six clinical isolates from the five patients shows that they differ by up to two single nucleotide polymorphisms, suggesting a common source of infection. Our findings are in accordance with the recognition of a new taxonomic entity. We propose the name M. basiliense, as all isolates were found in patients from the Basel area of Switzerland.
Keywords: Mycobacterium basiliense; nontuberculous mycobacteria; novel species; pathogen; virulence.
Figures
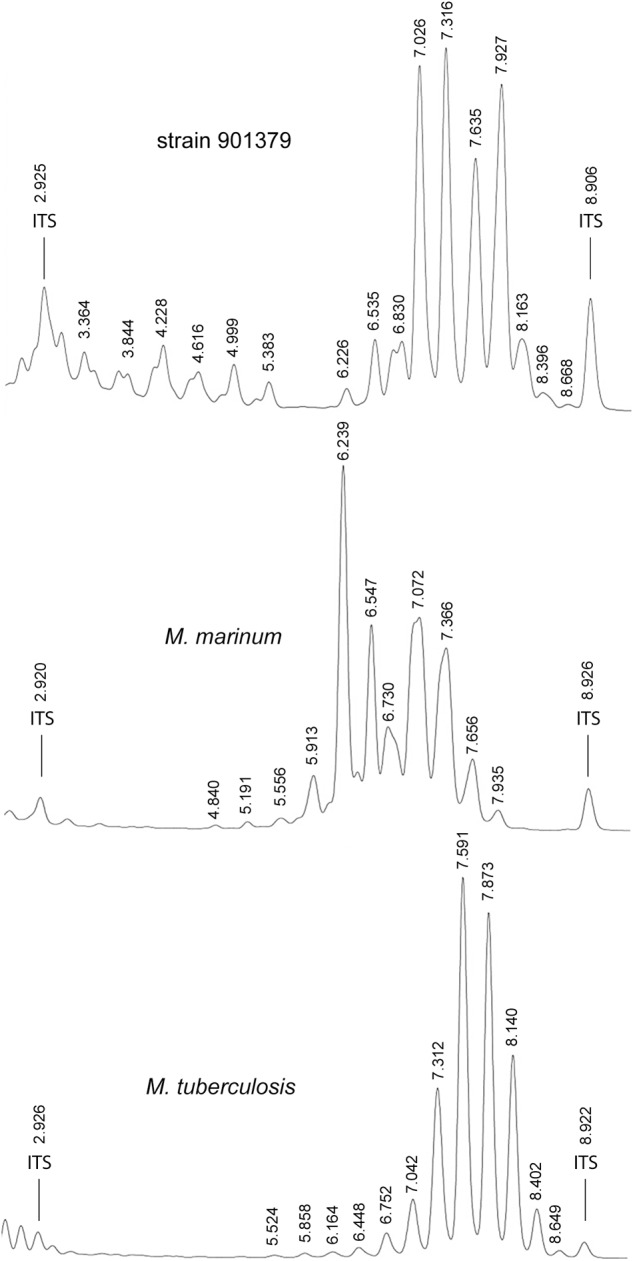

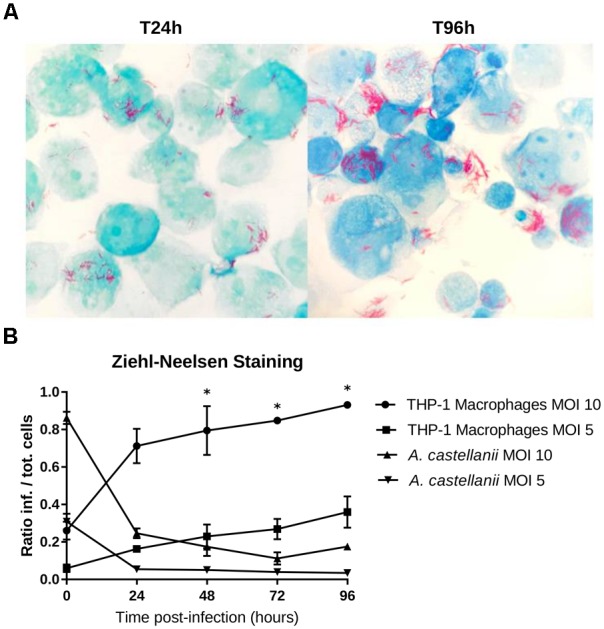
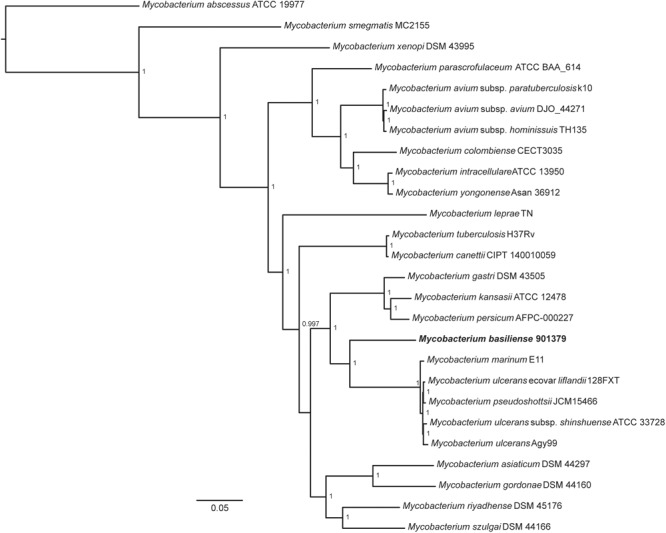
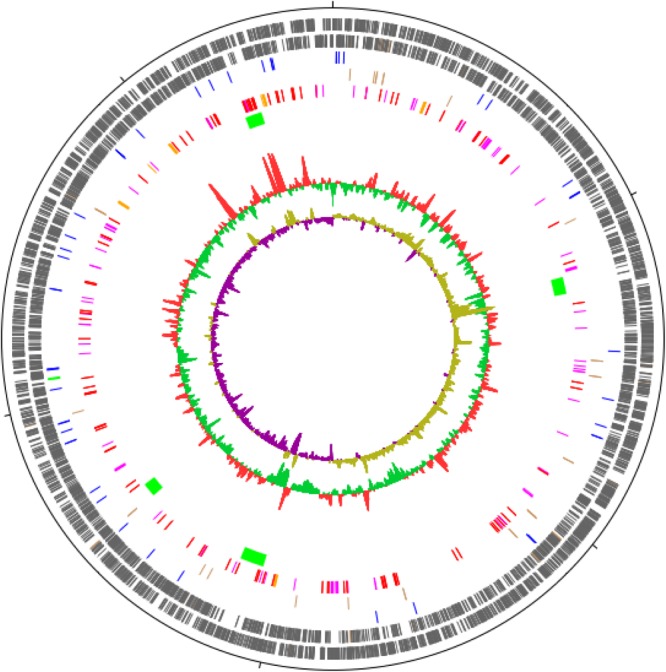

References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous