

The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses
- PMID: 30671058
- PMCID: PMC6332930
- DOI: 10.3389/fimmu.2018.03086
The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses
Abstract
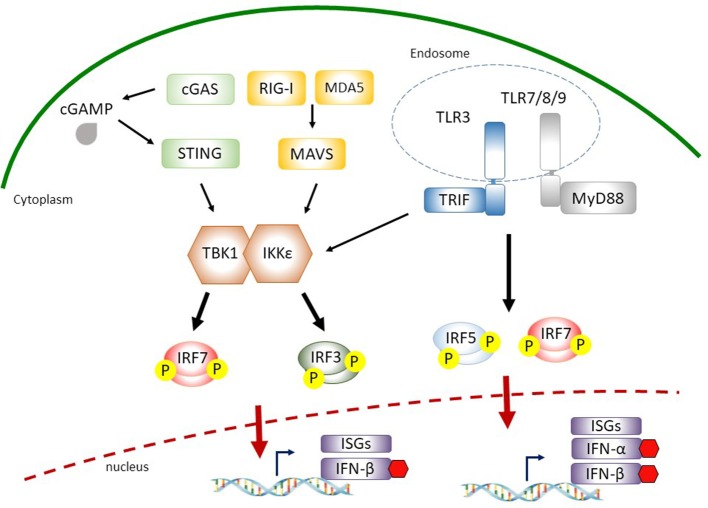
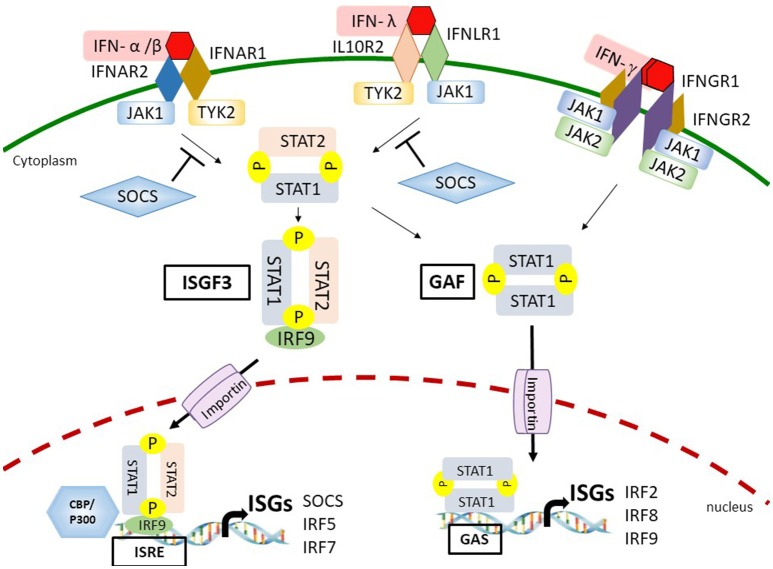
The antiviral innate immunity is the first line of host defense against virus infections. In mammalian cells, viral infections initiate the expression of interferons (IFNs) in the host that in turn activate an antiviral defense program to restrict viral replications by induction of IFN stimulated genes (ISGs), which are largely regulated by the IFN-regulatory factor (IRF) family and signal transducer and activator of transcription (STAT) family transcription factors. The mechanisms of action of IRFs and STATs involve several post-translational modifications, complex formation, and nuclear translocation of these transcription factors. However, many viruses, including human immunodeficiency virus (HIV), Zika virus (ZIKV), and herpes simplex virus (HSV), have evolved strategies to evade host defense, including alteration in IRF and STAT post-translational modifications, disturbing the formation and nuclear translocation of the transcription complexes as well as proteolysis/degradation of IRFs and STATs. In this review, we discuss and summarize the molecular mechanisms by which how viral components may target IRFs and STATs to antagonize the establishment of antiviral host defense. The underlying host-viral interactions determine the outcome of viral infection. Gaining mechanistic insight into these processes will be crucial in understanding how viral replication can be more effectively controlled and in developing approaches to improve virus infection outcomes.
Keywords: antiviral response; interferon; interferon-regulatory factor; interferon-stimulated gene; signal transducer and activator of transcription signaling; viral antagonism; viral attenuation.
Figures
Similar articles
-
IRF and STAT Transcription Factors - From Basic Biology to Roles in Infection, Protective Immunity, and Primary Immunodeficiencies.Front Immunol. 2019 Jan 8;9:3047. doi: 10.3389/fimmu.2018.03047. eCollection 2018. Front Immunol. 2019. PMID: 30671054 Free PMC article. Review.
-
Herpes Simplex Virus 1 UL36USP Antagonizes Type I Interferon-Mediated Antiviral Innate Immunity.J Virol. 2018 Sep 12;92(19):e01161-18. doi: 10.1128/JVI.01161-18. Print 2018 Oct 1. J Virol. 2018. PMID: 29997210 Free PMC article.
-
Pseudorabies Virus Inhibits Type I and Type III Interferon-Induced Signaling via Proteasomal Degradation of Janus Kinases.J Virol. 2021 Sep 27;95(20):e0079321. doi: 10.1128/JVI.00793-21. Epub 2021 Aug 11. J Virol. 2021. PMID: 34379505 Free PMC article.
-
Interference without interferon: interferon-independent induction of interferon-stimulated genes and its role in cellular innate immunity.mBio. 2024 Oct 16;15(10):e0258224. doi: 10.1128/mbio.02582-24. Epub 2024 Sep 20. mBio. 2024. PMID: 39302126 Free PMC article. Review.
-
Enterovirus 2Apro Cleavage of the YTHDF m6A Readers Implicates YTHDF3 as a Mediator of Type I Interferon-Driven JAK/STAT Signaling.mBio. 2021 Apr 13;12(2):e00116-21. doi: 10.1128/mBio.00116-21. mBio. 2021. PMID: 33849973 Free PMC article.
Cited by
-
Of Keeping and Tipping the Balance: Host Regulation and Viral Modulation of IRF3-Dependent IFNB1 Expression.Viruses. 2020 Jul 7;12(7):733. doi: 10.3390/v12070733. Viruses. 2020. PMID: 32645843 Free PMC article. Review.
-
Statistical and network analyses reveal mechanisms for the enhancement of macrophage immunity by manganese in Mycobacterium tuberculosis infection.Biochem Biophys Rep. 2023 Dec 20;37:101602. doi: 10.1016/j.bbrep.2023.101602. eCollection 2024 Mar. Biochem Biophys Rep. 2023. PMID: 38155943 Free PMC article.
-
Integration of Transcriptome and Proteome in Lymph Nodes Reveal the Different Immune Responses to PRRSV Between PRRSV-Resistant Tongcheng Pigs and PRRSV-Susceptible Large White Pigs.Front Genet. 2022 Jan 27;13:800178. doi: 10.3389/fgene.2022.800178. eCollection 2022. Front Genet. 2022. PMID: 35154273 Free PMC article.
-
Characteristics of herpes simplex virus infection and pathogenesis suggest a strategy for vaccine development.Rev Med Virol. 2019 Jul;29(4):e2054. doi: 10.1002/rmv.2054. Epub 2019 Jun 13. Rev Med Virol. 2019. PMID: 31197909 Free PMC article. Review.
-
Translational control of coronaviruses.Nucleic Acids Res. 2020 Dec 16;48(22):12502-12522. doi: 10.1093/nar/gkaa1116. Nucleic Acids Res. 2020. PMID: 33264393 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical