

Oncogenic Metabolism Acts as a Prerequisite Step for Induction of Cancer Metastasis and Cancer Stem Cell Phenotype
- PMID: 30671168
- PMCID: PMC6323533
- DOI: 10.1155/2018/1027453
Oncogenic Metabolism Acts as a Prerequisite Step for Induction of Cancer Metastasis and Cancer Stem Cell Phenotype
Abstract
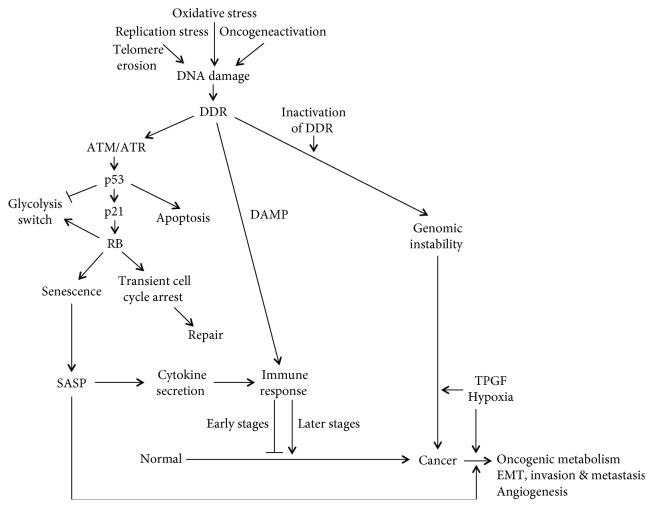
Metastasis is a major obstacle to the efficient and successful treatment of cancer. Initiation of metastasis requires epithelial-mesenchymal transition (EMT) that is regulated by several transcription factors, including Snail and ZEB1/2. EMT is closely linked to the acquisition of cancer stem cell (CSC) properties and chemoresistance, which contribute to tumor malignancy. Tumor suppressor p53 inhibits EMT and metastasis by negatively regulating several EMT-inducing transcription factors and regulatory molecules; thus, its inhibition is crucial in EMT, invasion, metastasis, and stemness. Metabolic alterations are another hallmark of cancer. Most cancer cells are more dependent on glycolysis than on mitochondrial oxidative phosphorylation for their energy production, even in the presence of oxygen. Cancer cells enhance other oncogenic metabolic pathways, such as glutamine metabolism, pentose phosphate pathway, and the synthesis of fatty acids and cholesterol. Metabolic reprogramming in cancer is regulated by the activation of oncogenes or loss of tumor suppressors that contribute to tumor progression. Oncogenic metabolism has been recently linked closely with the induction of EMT or CSC phenotypes by the induction of several metabolic enzyme genes. In addition, several transcription factors and molecules involved in EMT or CSCs, including Snail, Dlx-2, HIF-1α, STAT3, TGF-β, Wnt, and Akt, regulate oncogenic metabolism. Moreover, p53 induces metabolic change by directly regulating several metabolic enzymes. The collective data indicate the importance of oncogenic metabolism in the regulation of EMT, cell invasion and metastasis, and adoption of the CSC phenotype, which all contribute to malignant transformation and tumor development. In this review, we highlight the oncogenic metabolism as a key regulator of EMT and CSC, which is related with tumor progression involving metastasis and chemoresistance. Targeting oncometabolism might be a promising strategy for the development of effective anticancer therapy.
Figures

Similar articles
-
Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation.Mol Cancer. 2017 Jan 30;16(1):10. doi: 10.1186/s12943-016-0577-4. Mol Cancer. 2017. PMID: 28137309 Free PMC article. Review.
-
Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling.J Hematol Oncol. 2015 Mar 11;8:23. doi: 10.1186/s13045-015-0119-3. J Hematol Oncol. 2015. PMID: 25879771 Free PMC article.
-
Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition.Cells. 2020 Jan 15;9(1):217. doi: 10.3390/cells9010217. Cells. 2020. PMID: 31952344 Free PMC article. Review.
-
HIF-1α induces the epithelial-mesenchymal transition in gastric cancer stem cells through the Snail pathway.Oncotarget. 2017 Feb 7;8(6):9535-9545. doi: 10.18632/oncotarget.14484. Oncotarget. 2017. PMID: 28076840 Free PMC article.
-
Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch.Oncotarget. 2016 Feb 16;7(7):7925-39. doi: 10.18632/oncotarget.6879. Oncotarget. 2016. PMID: 26771232 Free PMC article.
Cited by
-
P53 together with ferroptosis: a promising strategy leaving cancer cells without escape.Acta Biochim Biophys Sin (Shanghai). 2024 Jan 25;56(1):1-14. doi: 10.3724/abbs.2023270. Acta Biochim Biophys Sin (Shanghai). 2024. PMID: 38105650 Free PMC article.
-
Potential role of epithelial-mesenchymal transition induced by periodontal pathogens in oral cancer.J Cell Mol Med. 2024 Jan;28(1):e18064. doi: 10.1111/jcmm.18064. Epub 2023 Nov 30. J Cell Mol Med. 2024. PMID: 38031653 Free PMC article. Review.
-
Correlation between amino acid metabolism and self-renewal of cancer stem cells: Perspectives in cancer therapy.World J Stem Cells. 2022 Apr 26;14(4):267-286. doi: 10.4252/wjsc.v14.i4.267. World J Stem Cells. 2022. PMID: 35662861 Free PMC article. Review.
-
Dual functionality of MDM2 in PROTACs expands the horizons of targeted protein degradation.Biomark Res. 2025 Aug 27;13(1):111. doi: 10.1186/s40364-025-00826-7. Biomark Res. 2025. PMID: 40866949 Free PMC article. Review.
-
A Shifty Target: Tumor-Initiating Cells and Their Metabolism.Int J Mol Sci. 2019 Oct 28;20(21):5370. doi: 10.3390/ijms20215370. Int J Mol Sci. 2019. PMID: 31661927 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
