

Review
doi: 10.1056/NEJMra1800222.
Opioid Tolerance in Critical Illness
Affiliations
- PMID: 30673555
- PMCID: PMC6897318
- DOI: 10.1056/NEJMra1800222
Item in Clipboard
Review
Opioid Tolerance in Critical Illness
N Engl J Med.
.
No abstract available
Figures
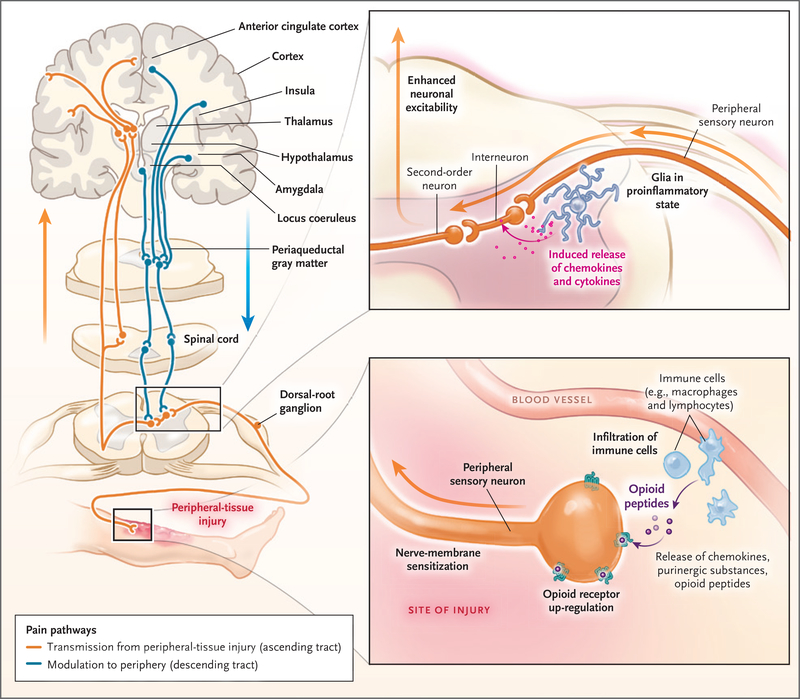
Sites of action of opioids for pain relief include the brain (cortex, thalamus, hypothalamus, locus coeruleus, amygdala, and periaqueductal gray matter), spinal cord, and peripheral-nerve membrane. Transmission of pain sensation (nociception) from the peripheral-tissue injury to the central nervous system occurs through the ascending spinothalamic tract to the thalamus and then to the somatosensory cortex (orange). Descending inhibitory tracts (blue) from the brain and other regions, including the rostroventral medulla, modulate nociception. Nociception can be amplified by dorsal-root ganglia and changes in the dorsal horn of the spinal cord (top inset). The afferent neurons are sensitized by the sprouting of new axons around the cell bodies of dorsal-root ganglia, as well as by infiltrating macrophages, which release inflammatory substances. Neuron projections from dorsal-root ganglia to the dorsal horn amplify the pain by the release of other pro-nociceptive mediators (e.g., calcitonin gene–related peptide), activation of N-methyl-d -aspartate receptors, and the increase in glutamate levels. Second-order neurons transmit these signals upstream to the brain (orange). Injury to tissues (bottom inset) results in local and often systemic inflammatory responses, which prime the peripheral sensory neurons and dorsal-root ganglia to exaggerated nociception by up-regulation or modulation of ligand-gated and voltage-gated ion channels. Mu-opioid receptors are newly expressed throughout the nerve membrane. Extravasated circulating leukocytes (e.g., macrophages and lymphocytes) release proinflammatory mediators, further sensitizing the neurons to pain. These leukocytes also release antinociceptive endogenous opioid peptides, which bind to the up-regulated opioid receptors on the nerve, attenuating pain.
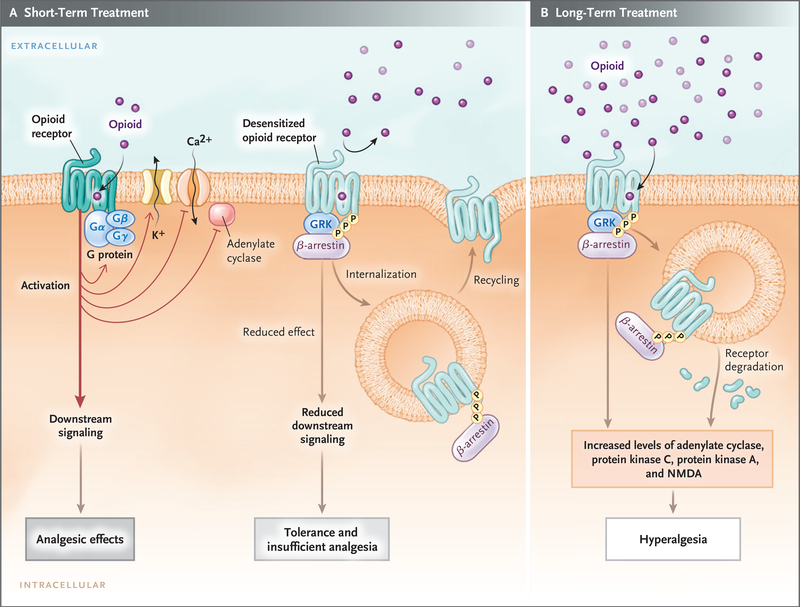
In short-term treatment, the binding of an opioid to its receptor (Panel A) causes downstream G-protein–coupled receptors, composed of Gαβγ subunits, to dissociate into Gα and Gβγ subunits. The dissociated G-protein subunits inhibit voltage-gated calcium channels by means of reduced transmitter release, activate inward-rectifying potassium channels (causing hyperpolarization of the membrane), and inhibit downstream adenylate cyclase enzymes, decreasing cyclic adenosine monophosphate levels. These events reduce excitability and nociception and result in analgesic effects. When an opioid binds to its receptor, it becomes an immediate substrate for phosphorylation by G-protein–coupled receptor kinase (GRK), which leads to recruitment and binding of β-arrestin protein to the receptor. This results in desensitization and sometimes endocytosis of the receptor; each of these events decreases the responses to opioids, inducing tolerance and insufficient analgesia. Opioid-receptor signaling terminates when the opioid is displaced from the receptor. After the stimulus (i.e., the agonist) is withdrawn, the desensitized receptor recovers over time (minutes to hours, depending on the agonist), Gα rebinds to Gβγ and once again forms Gαβγ, and the endocytosed receptor is reexpressed on the plasma membrane in a resensitized state. In long-term treatment (Panel B), escalating doses of opioids and concomitant persistent activation of the receptor lead to aggravation of the tolerance by receptor-dependent and receptor-independent intracellular signaling changes, which include up-regulation of the antiopioid (pro-nociceptive) signaling pathways. The sustained β-arrestin binding to the receptor often leads to internalization, degradation, and down-regulation of membrane receptor number, further decreasing response to opioids. Receptor down-regulation occurs with some opioids (e.g., fentanyl) but not others (e.g., morphine). Phosphorylation by other kinases (e.g., protein kinases A and C), increased adenylate cyclase activity (with increased cyclic adenosine monophosphate levels), activation of N-methyl-d -aspartate (NMDA) receptor, and down-regulation of glutamate receptors (increased glutamate levels) are all implicated in the imbalance between pro-nociceptive and antinociceptive pathways, which results in attenuated analgesic effects, aggravated pain behaviors, increased tolerance, and opioid-induced hyperalgesia.
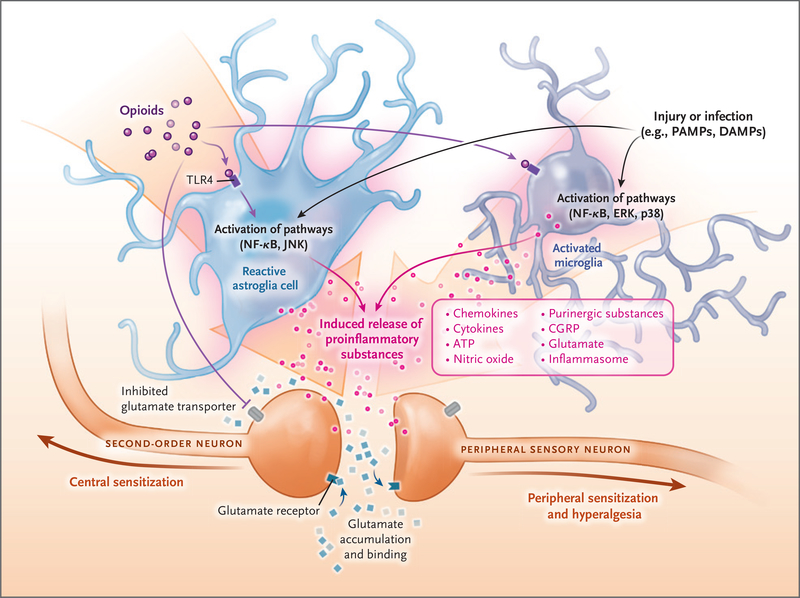
Non-neuronal cells (e.g., astroglia and microglia) can modify pain perception through the production and release of pro-nociceptive m ediators. Opioids, injury, cancer, chemotherapy, stress, and other causes of sterile or microbial inflammation can induce the release of damage-associated molecular pathogens (DAMPs) and pathogen-associated molecular patterns (PAMPs). DAMPs and PAMPs cause inflammasome release, which leads to the transition of microglia to an active state and astroglia to a reactive state. The “switched on” glia release inflammatory substances through activation of toll-like receptors (TLRs) and their downstream signaling proteins (Jun N-terminal kinase [JNK], nuclear factor κB [NF-κB], extracellular signal-regulated kinase [ERK], and p38 mitogen-activated protein kinase [p38]). Peripheral macrophages infiltrate the central nervous system because of selective breakdown of the blood–brain barrier, and they contribute to the inflammatory responses. The released proinflammatory substances (inflammasomes, ATP, and calcitonin gene–related peptide [CGRP]) sensitize the pre- and postsynaptic central neurons, leading to a vicious cycle characterized by the need for more opioids and more sensitization and more glia inflammation. The end result is a marked exaggeration of nociception, severe opioid tolerance, peripheral and central sensitization, and opioid-induced hyperalgesia.
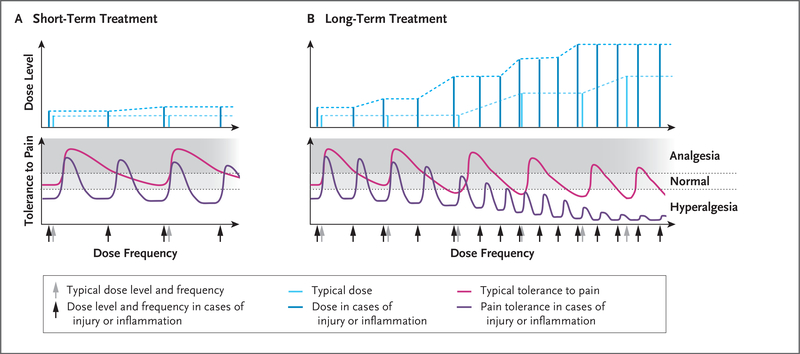
In Panel A, short-term opioid administration (light blue) provides sufficient analgesia with no or minimal opioid tolerance (pink). In cases of inflammation or injury, as compared with uninjured states, the analgesic potency of the opioid (i.e., the threshold for pain) is decreased, resulting in hyperalgesia; short-term administration of higher doses (dark blue) provides sufficient analgesia but for a shorter duration (purple), requiring more frequent doses (dark blue). Panel B shows that in an uninjured patient with long-term exposure to opioids, the analgesic potency decreases (pink) and the duration of opioid-induced analgesia also decreases with each dose, requiring an increase in dose frequency. Long-term opioid administration will result in induction of antinociceptive mechanisms, resulting in hyperalgesia; even higher doses of opioids (light blue) do not restore complete analgesia (pink). During opioid-induced hyperalgesia in an injured patient, exaggerated pain sensitivity occurs at the injured and uninjured areas. Any cause of systemic inflammation or neuroinflammation (e.g., infection, cancer, diabetes, stress, or chemotherapy) decreases the analgesic potency and the duration of analgesic effects and leads to earlier development of opioid-induced hyperalgesia; even high doses (dark blue) result in minimal analgesia (purple) because of decreased analgesic potency.
Comment in
-
Opioid Tolerance in Critical Illness.N Engl J Med. 2019 Apr 18;380(16):e26. doi: 10.1056/NEJMc1902646. N Engl J Med. 2019. PMID: 30995389 No abstract available.
References
-
- Cullen DJ, Sweitzer BJ, Bates DW, Burdick E, Edmondson A, Leape LL. Preventable adverse drug events in hospitalized patients: a comparative study of intensive care and general care units. Crit Care Med 1997; 25: 1289–97. - PubMed
-
- Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013; 41: 263–306. - PubMed
-
- Roeckel LA, Le Coz GM, Gavériaux-Ruff C, Simonin F. Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience 2016; 338:1 60–82. - PubMed
-
- Chu LF, Angst MS, Clark D. Opioid-induced hyperalgesia in humans: molecular mechanisms and clinical considerations. Clin J Pain 2008; 24: 479–96. - PubMed
-
- Puntillo KA, Naidu R. Chronic pain disorders after critical illness and ICU-acquired opioid dependence: two clinical conundra. Curr Opin Crit Care 2016; 22: 506–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical