

Evolutionary dynamics of bacteria in the gut microbiome within and across hosts
- PMID: 30673701
- PMCID: PMC6361464
- DOI: 10.1371/journal.pbio.3000102
Evolutionary dynamics of bacteria in the gut microbiome within and across hosts
Abstract
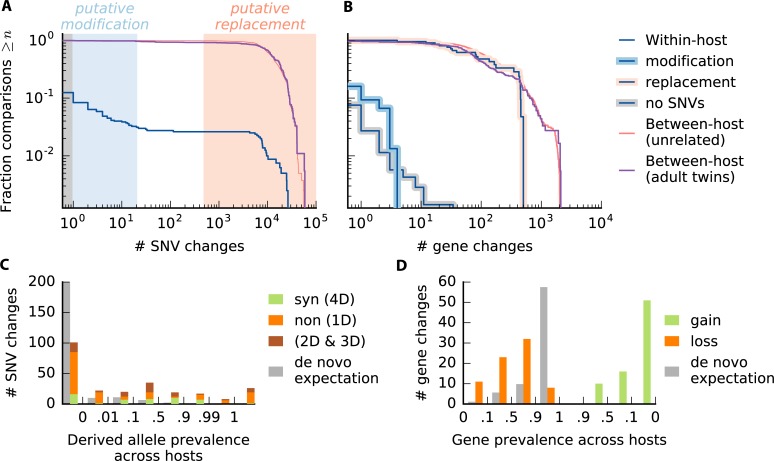
Gut microbiota are shaped by a combination of ecological and evolutionary forces. While the ecological dynamics have been extensively studied, much less is known about how species of gut bacteria evolve over time. Here, we introduce a model-based framework for quantifying evolutionary dynamics within and across hosts using a panel of metagenomic samples. We use this approach to study evolution in approximately 40 prevalent species in the human gut. Although the patterns of between-host diversity are consistent with quasi-sexual evolution and purifying selection on long timescales, we identify new genealogical signatures that challenge standard population genetic models of these processes. Within hosts, we find that genetic differences that accumulate over 6-month timescales are only rarely attributable to replacement by distantly related strains. Instead, the resident strains more commonly acquire a smaller number of putative evolutionary changes, in which nucleotide variants or gene gains or losses rapidly sweep to high frequency. By comparing these mutations with the typical between-host differences, we find evidence that some sweeps may be seeded by recombination, in addition to new mutations. However, comparisons of adult twins suggest that replacement eventually overwhelms evolution over multi-decade timescales, hinting at fundamental limits to the extent of local adaptation. Together, our results suggest that gut bacteria can evolve on human-relevant timescales, and they highlight the connections between these short-term evolutionary dynamics and longer-term evolution across hosts.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
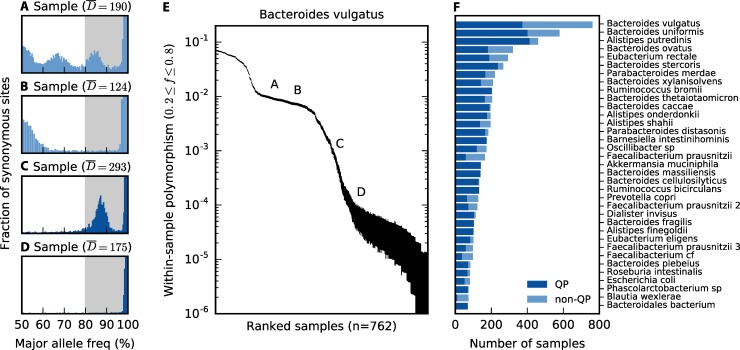
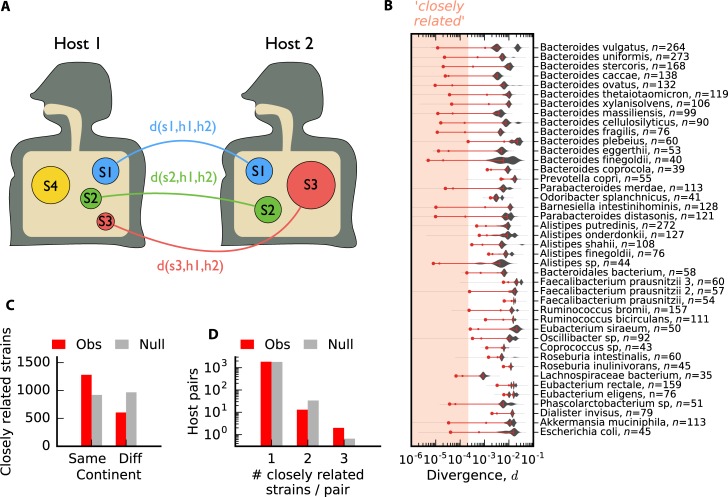
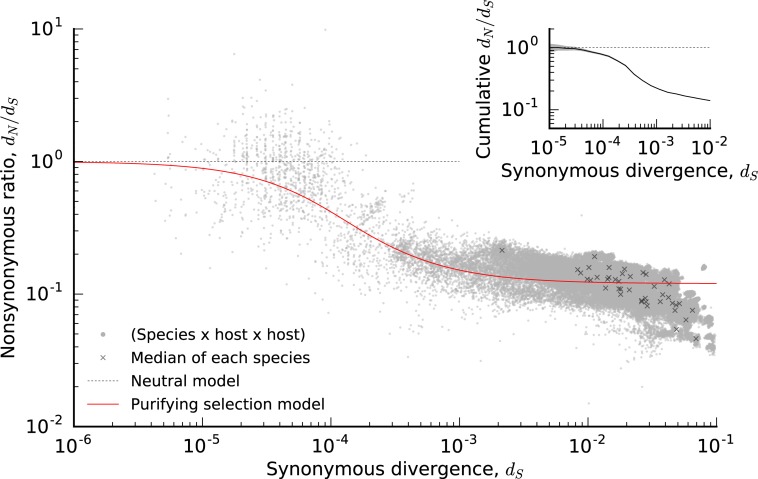
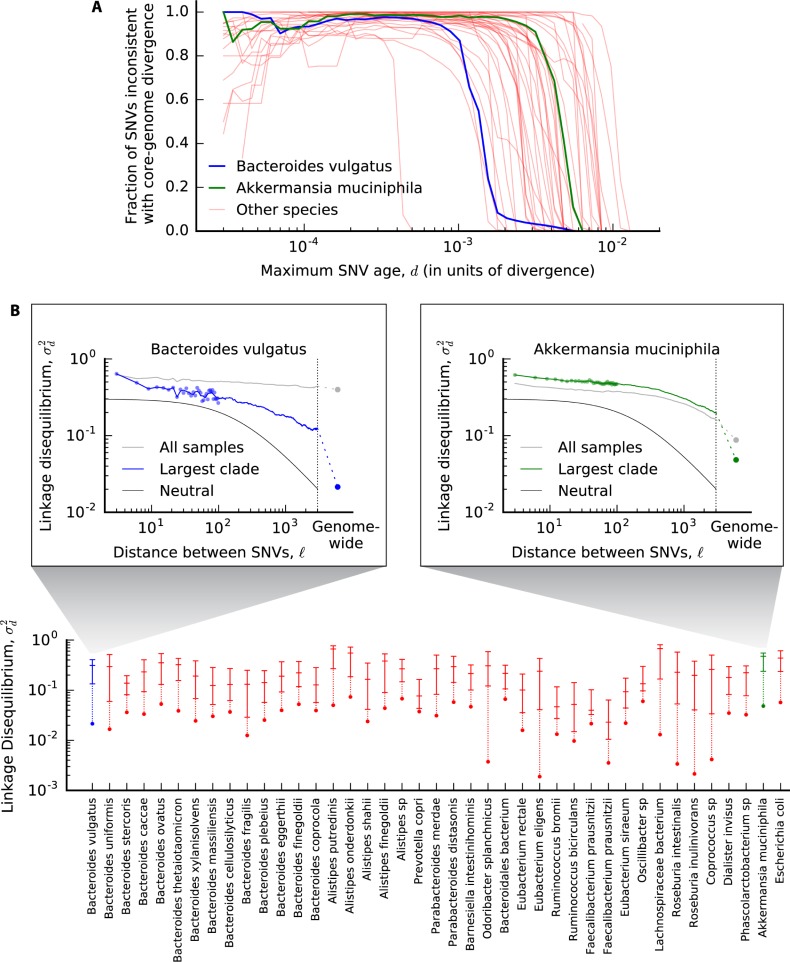
Comment in
-
Evolutionary change in the human gut microbiome: From a static to a dynamic view.PLoS Biol. 2019 Feb 7;17(2):e3000126. doi: 10.1371/journal.pbio.3000126. eCollection 2019 Feb. PLoS Biol. 2019. PMID: 30730933 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
