

Mitochondrial stress triggers a pro-survival response through epigenetic modifications of nuclear DNA
- PMID: 30673822
- PMCID: PMC11105675
- DOI: 10.1007/s00018-019-03008-5
Mitochondrial stress triggers a pro-survival response through epigenetic modifications of nuclear DNA
Abstract
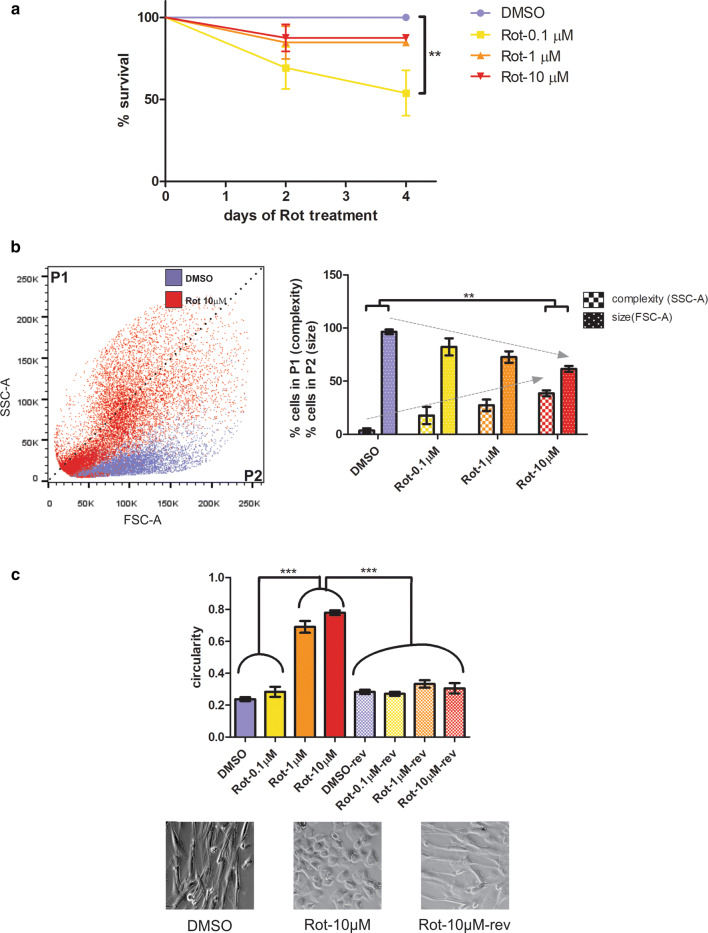
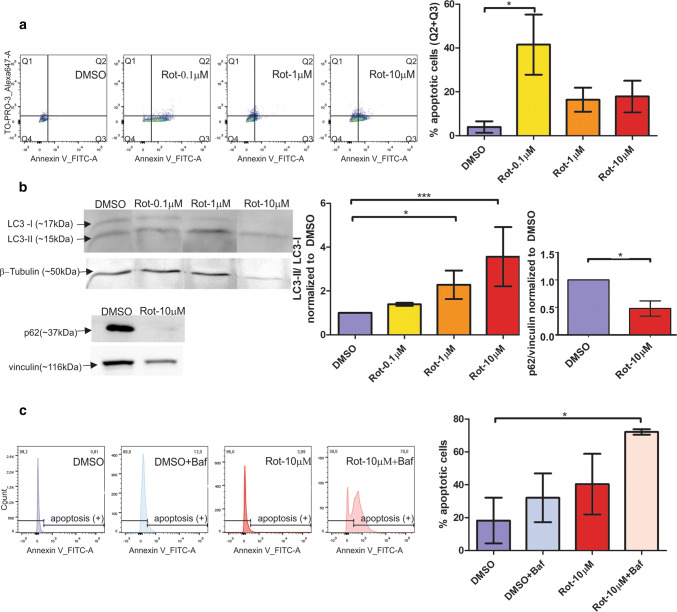
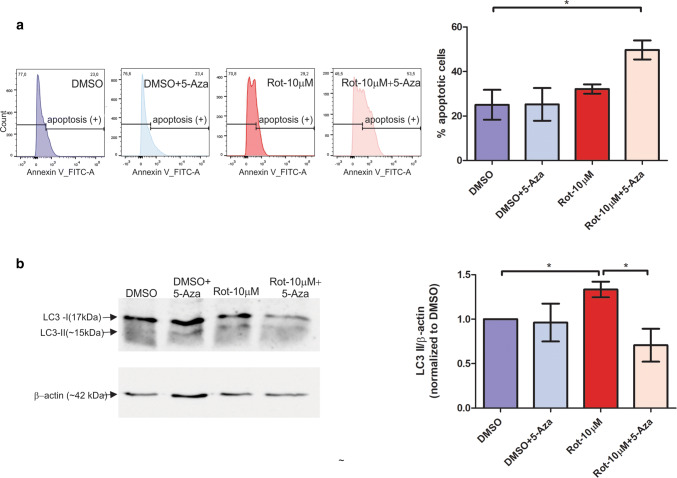
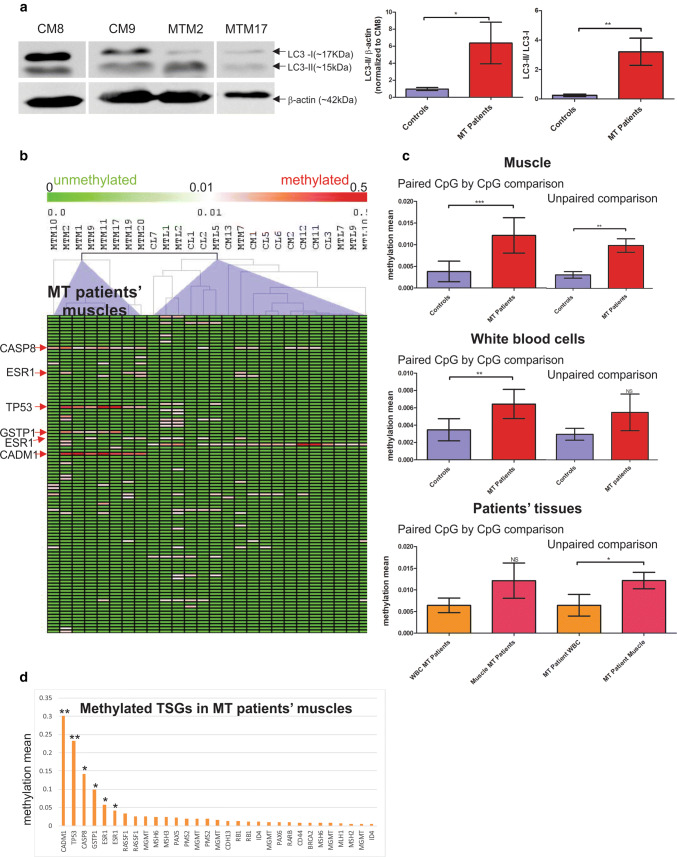
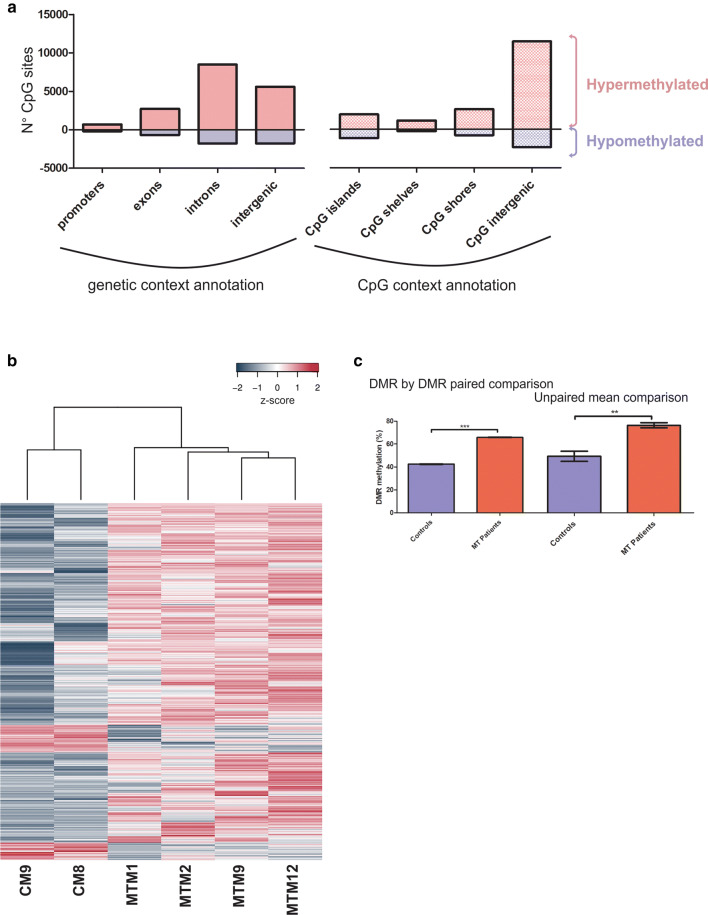
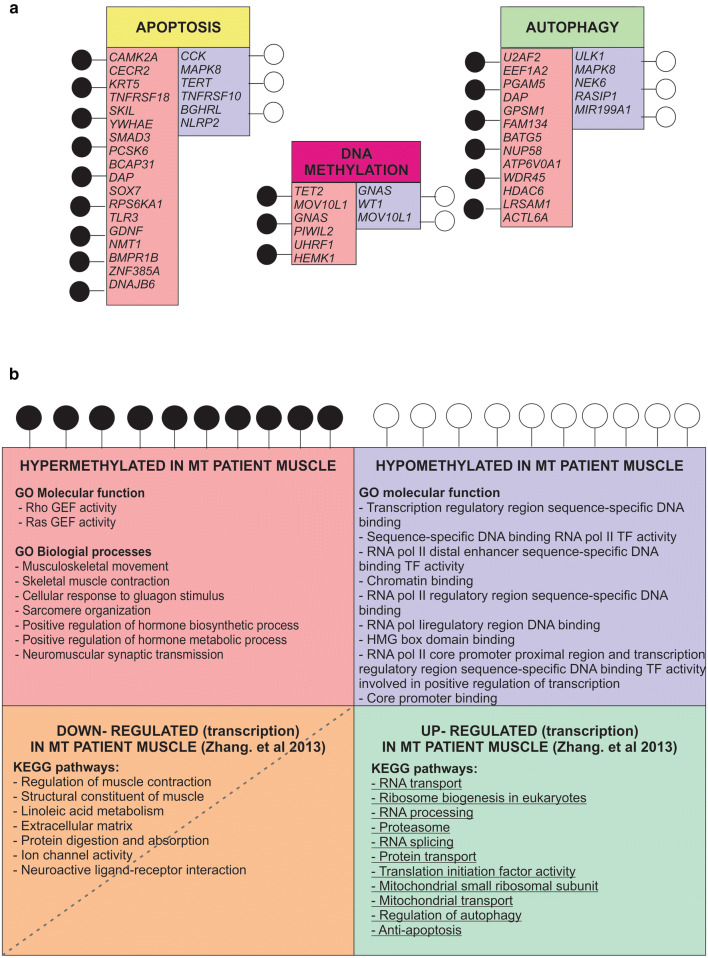
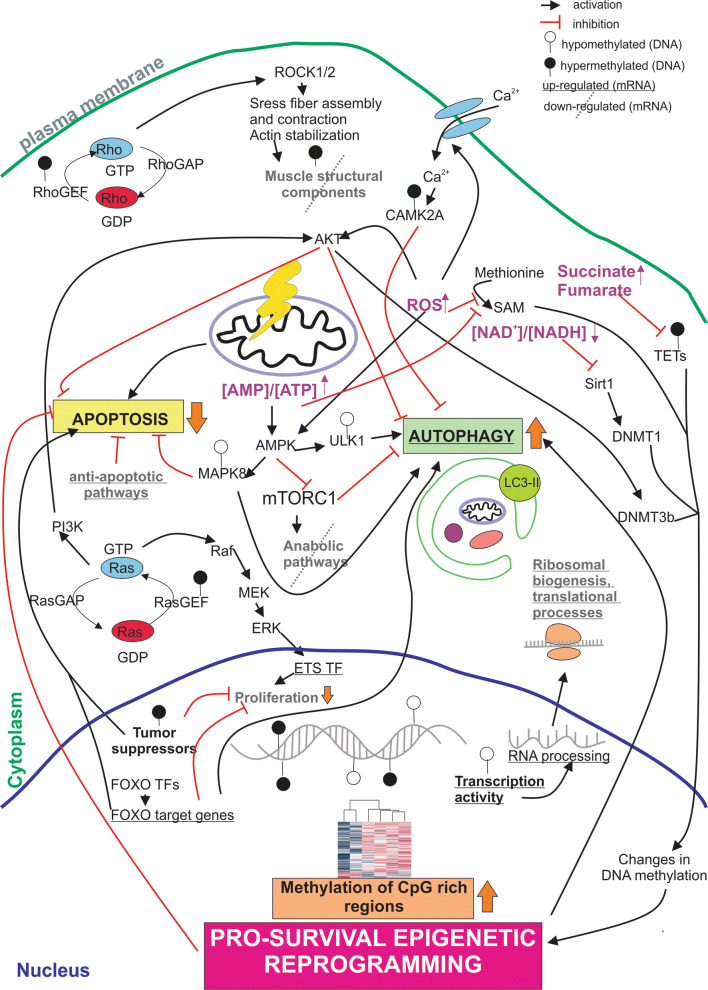
Mitochondrial dysfunction represents an important cellular stressor and when intense and persistent cells must unleash an adaptive response to prevent their extinction. Furthermore, mitochondria can induce nuclear transcriptional changes and DNA methylation can modulate cellular responses to stress. We hypothesized that mitochondrial dysfunction could trigger an epigenetically mediated adaptive response through a distinct DNA methylation patterning. We studied cellular stress responses (i.e., apoptosis and autophagy) in mitochondrial dysfunction models. In addition, we explored nuclear DNA methylation in response to this stressor and its relevance in cell survival. Experiments in cultured human myoblasts revealed that intense mitochondrial dysfunction triggered a methylation-dependent pro-survival response. Assays done on mitochondrial disease patient tissues showed increased autophagy and enhanced DNA methylation of tumor suppressor genes and pathways involved in cell survival regulation. In conclusion, mitochondrial dysfunction leads to a "pro-survival" adaptive state that seems to be triggered by the differential methylation of nuclear genes.
Keywords: Apoptosis; Autophagy; DNA methylation; Mitochondrial diseases; Mitochondrial dysfunction; Stress response; Survival.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Mitochondrial metabolism and DNA methylation: a review of the interaction between two genomes.Clin Epigenetics. 2020 Nov 23;12(1):182. doi: 10.1186/s13148-020-00976-5. Clin Epigenetics. 2020. PMID: 33228792 Free PMC article. Review.
-
Mitochondrial regulation of epigenetics and its role in human diseases.Epigenetics. 2012 Apr;7(4):326-34. doi: 10.4161/epi.19547. Epub 2012 Apr 1. Epigenetics. 2012. PMID: 22419065 Free PMC article. Review.
-
Mitochondrial DNA Methylation and Human Diseases.Int J Mol Sci. 2021 Apr 27;22(9):4594. doi: 10.3390/ijms22094594. Int J Mol Sci. 2021. PMID: 33925624 Free PMC article. Review.
-
Novel incretin analogues improve autophagy and protect from mitochondrial stress induced by rotenone in SH-SY5Y cells.J Neurochem. 2016 Oct;139(1):55-67. doi: 10.1111/jnc.13736. Epub 2016 Aug 24. J Neurochem. 2016. PMID: 27412483
-
The Role of N6-Methyladenosine in Mitochondrial Dysfunction and Pathology.Int J Mol Sci. 2025 Apr 11;26(8):3624. doi: 10.3390/ijms26083624. Int J Mol Sci. 2025. PMID: 40332101 Free PMC article. Review.
Cited by
-
Mitochondrial DNA methylation is involved in contrast-induced renal tubular epithelial cell injury.Ren Fail. 2025 Dec;47(1):2532112. doi: 10.1080/0886022X.2025.2532112. Epub 2025 Aug 10. Ren Fail. 2025. PMID: 40784878 Free PMC article.
-
The effect of mitochondrial transplantation therapy from different gender on inhibiting cell proliferation of malignant melanoma.Int J Biol Sci. 2021 May 11;17(8):2021-2033. doi: 10.7150/ijbs.59581. eCollection 2021. Int J Biol Sci. 2021. PMID: 34131403 Free PMC article.
-
Structure, mechanism, and regulation of mitochondrial DNA transcription initiation.J Biol Chem. 2020 Dec 25;295(52):18406-18425. doi: 10.1074/jbc.REV120.011202. Epub 2020 Oct 30. J Biol Chem. 2020. PMID: 33127643 Free PMC article. Review.
-
Ionizing radiations induce shared epigenomic signatures unraveling adaptive mechanisms of cancerous cell lines with or without methionine dependency.Clin Epigenetics. 2021 Dec 1;13(1):212. doi: 10.1186/s13148-021-01199-y. Clin Epigenetics. 2021. PMID: 34852845 Free PMC article.
-
Rewriting nuclear epigenetic scripts in mitochondrial diseases as a strategy for heteroplasmy control.EMBO Mol Med. 2025 Aug 11. doi: 10.1038/s44321-025-00285-5. Online ahead of print. EMBO Mol Med. 2025. PMID: 40790102
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
