

Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin
- PMID: 30674652
- PMCID: PMC7184965
- DOI: 10.1126/scitranslmed.aat1199
Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin
Abstract
The mechanisms by which truncating mutations in MYBPC3 (encoding cardiac myosin-binding protein C; cMyBPC) or myosin missense mutations cause hypercontractility and poor relaxation in hypertrophic cardiomyopathy (HCM) are incompletely understood. Using genetic and biochemical approaches, we explored how depletion of cMyBPC altered sarcomere function. We demonstrated that stepwise loss of cMyBPC resulted in reciprocal augmentation of myosin contractility. Direct attenuation of myosin function, via a damaging missense variant (F764L) that causes dilated cardiomyopathy (DCM), normalized the increased contractility from cMyBPC depletion. Depletion of cMyBPC also altered dynamic myosin conformations during relaxation, enhancing the myosin state that enables ATP hydrolysis and thin filament interactions while reducing the super relaxed conformation associated with energy conservation. MYK-461, a pharmacologic inhibitor of myosin ATPase, rescued relaxation deficits and restored normal contractility in mouse and human cardiomyocytes with MYBPC3 mutations. These data define dosage-dependent effects of cMyBPC on myosin that occur across the cardiac cycle as the pathophysiologic mechanisms by which MYBPC3 truncations cause HCM. Therapeutic strategies to attenuate cMyBPC activity may rescue depressed cardiac contractility in patients with DCM, whereas inhibiting myosin by MYK-461 should benefit the substantial proportion of patients with HCM with MYBPC3 mutations.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Competing Financial Interest
C.E.S. and J.G.S. are founders and own shares in Myokardia Inc., a startup company that is developing therapeutics that target the sarcomere.
Figures
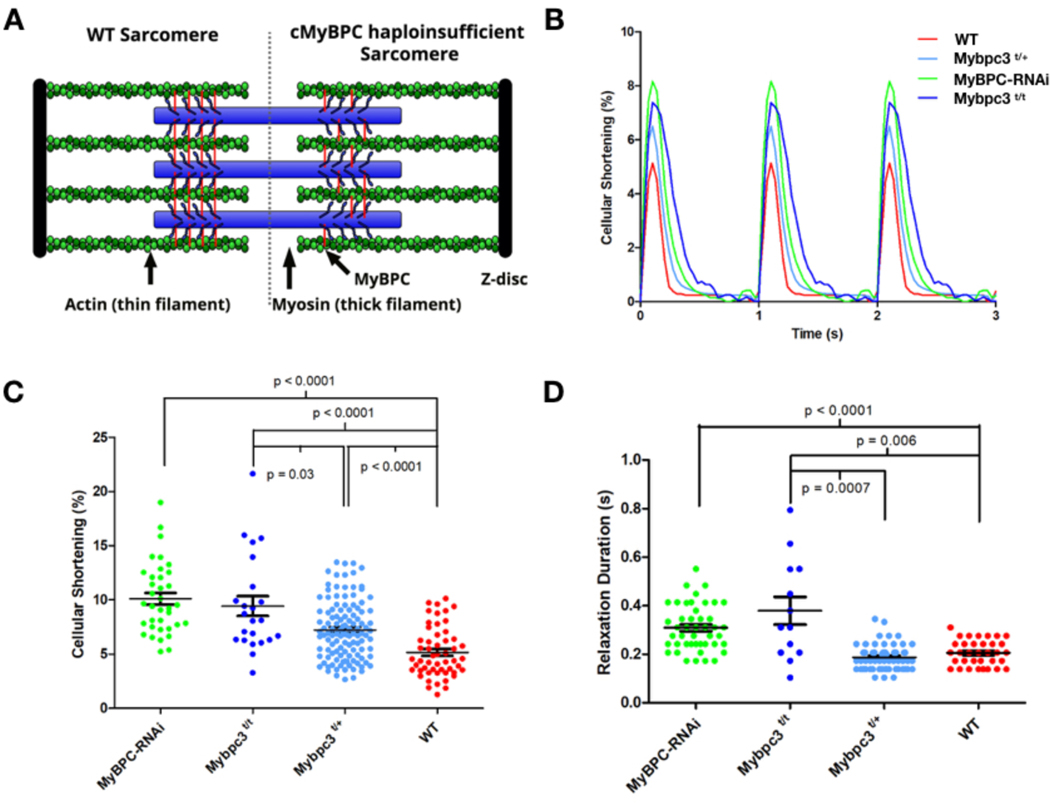
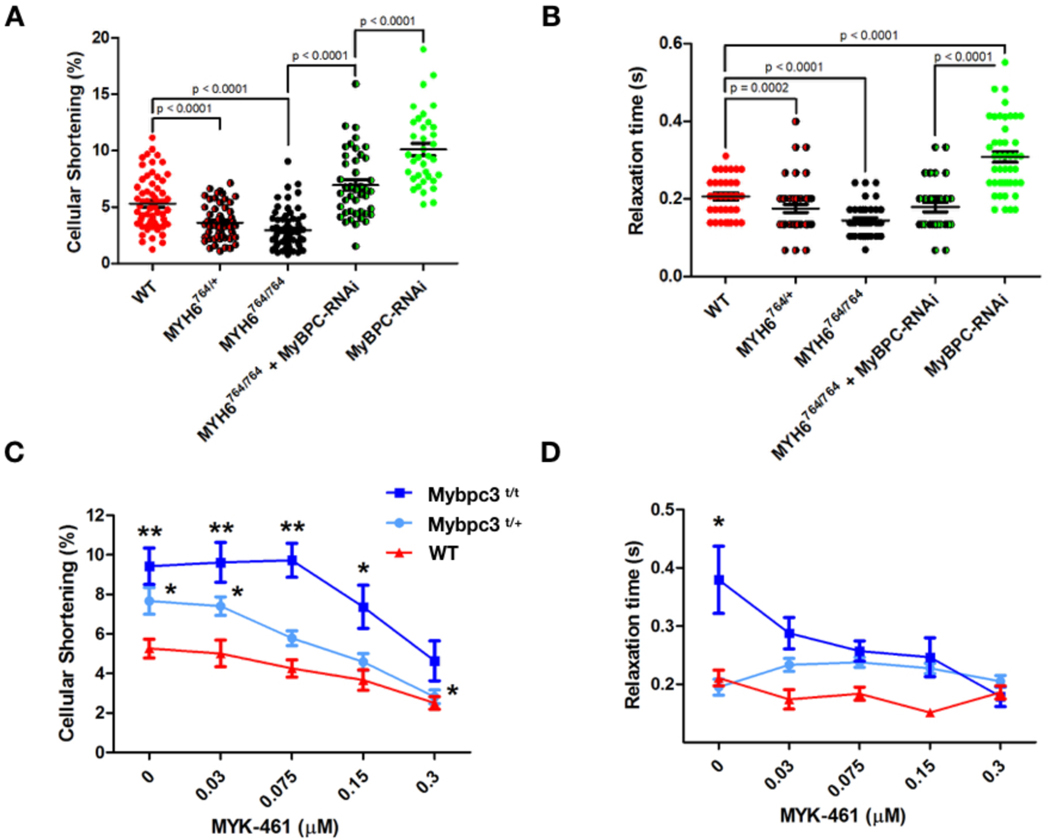
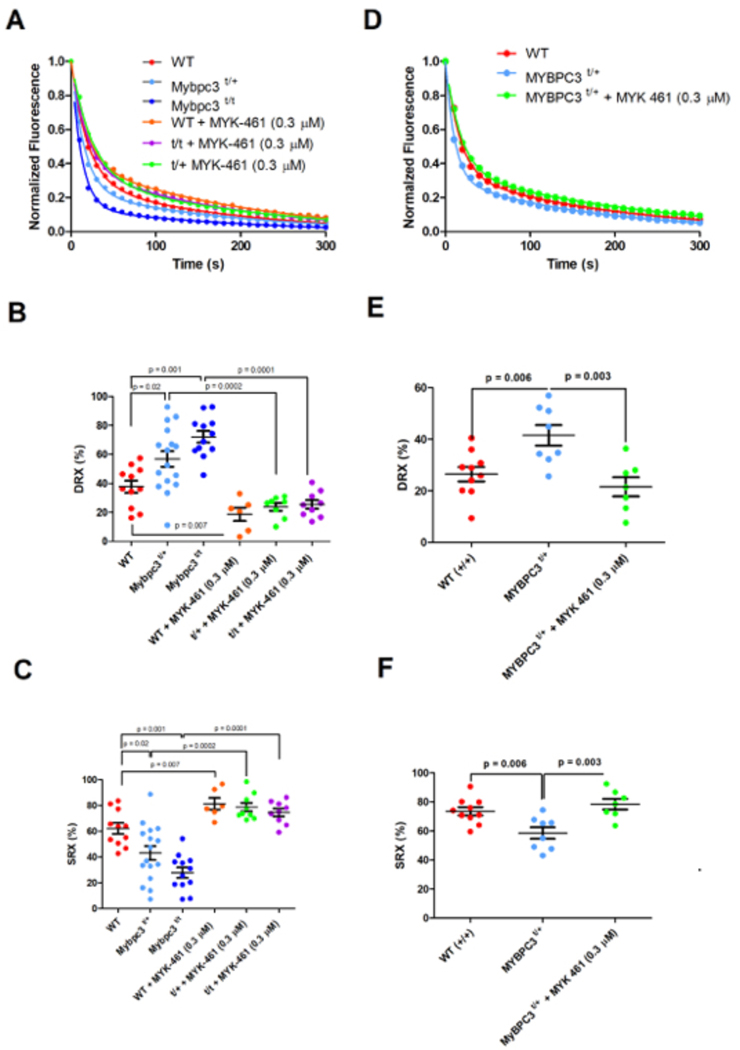
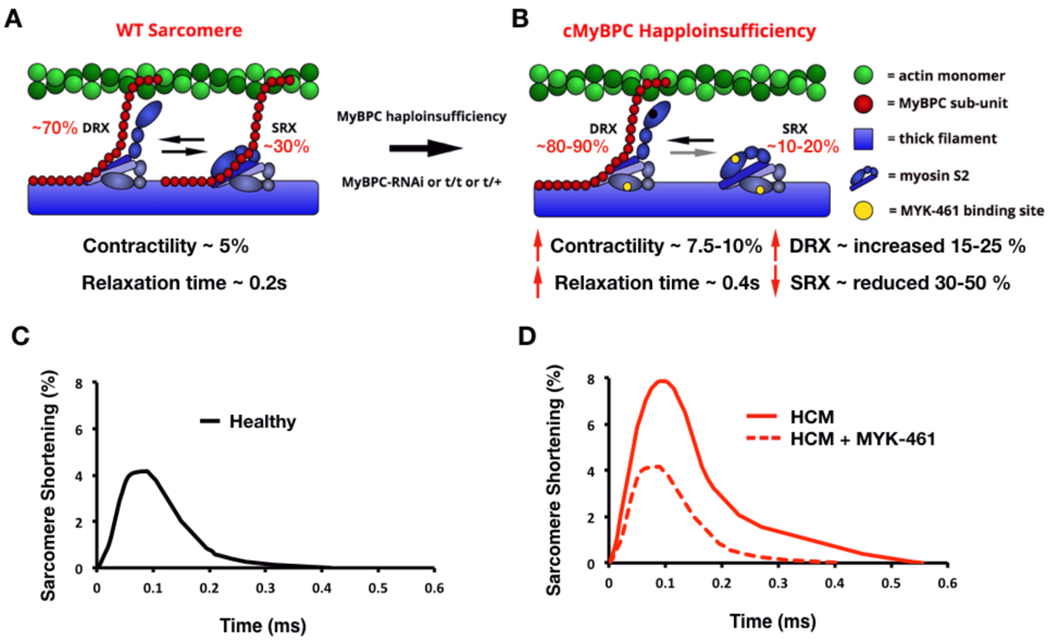
Comment in
-
Modulating myosin function to treat hypertrophic cardiomyopathy.Nat Rev Cardiol. 2019 Apr;16(4):201. doi: 10.1038/s41569-019-0170-9. Nat Rev Cardiol. 2019. PMID: 30755739 No abstract available.
References
-
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT and Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–9. - PubMed
-
- Maron BJ, Casey SA, Poliac LC, Gohman TE, Almquist AK and Aeppli DM. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650–5. - PubMed
-
- Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG and Seidman CE. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–21. - PMC - PubMed
-
- Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Exome Aggregation C, MacArthur DG, Farrall M, Cook SA and Watkins H. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
