

Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model
- PMID: 30674713
- PMCID: PMC6413832
- DOI: 10.1172/jci.insight.125067
Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model
Abstract
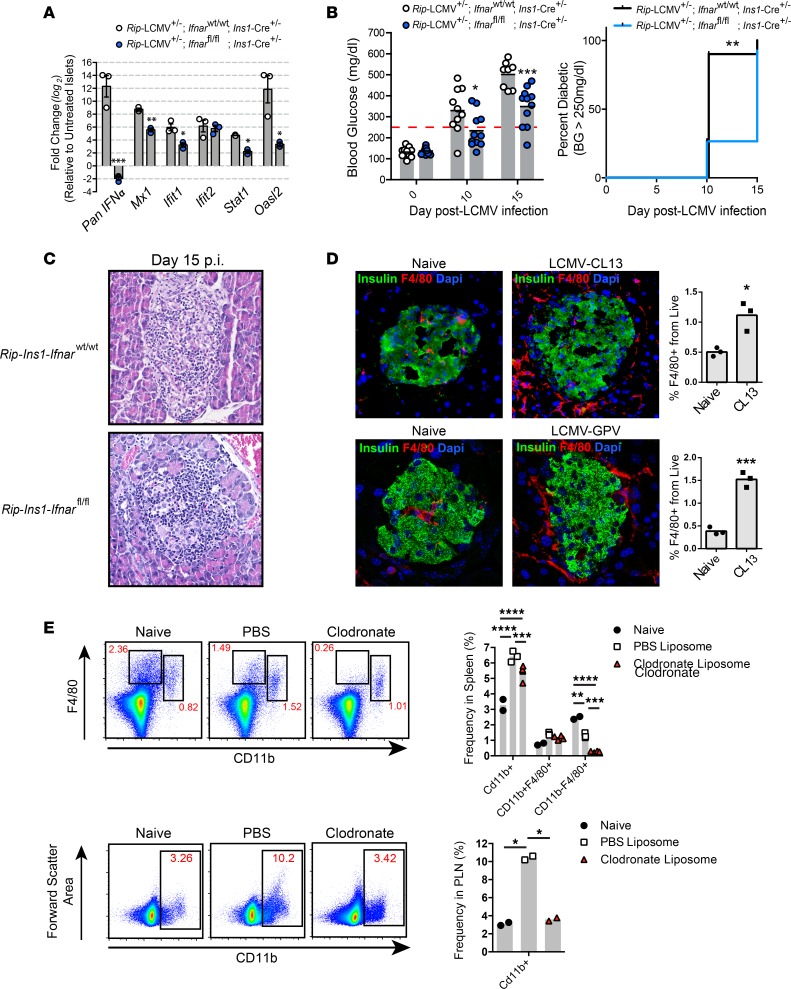
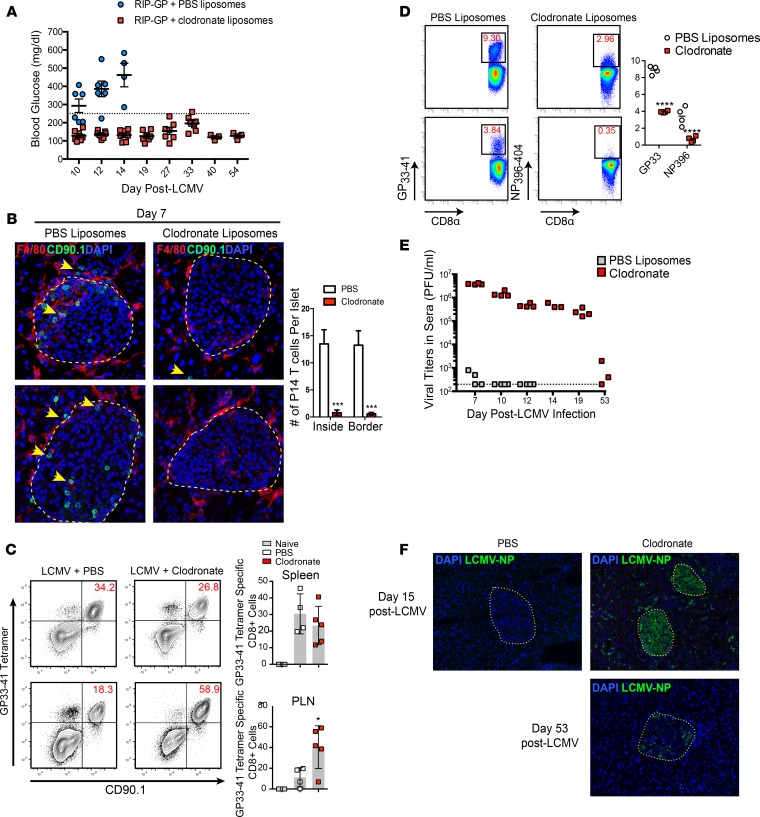
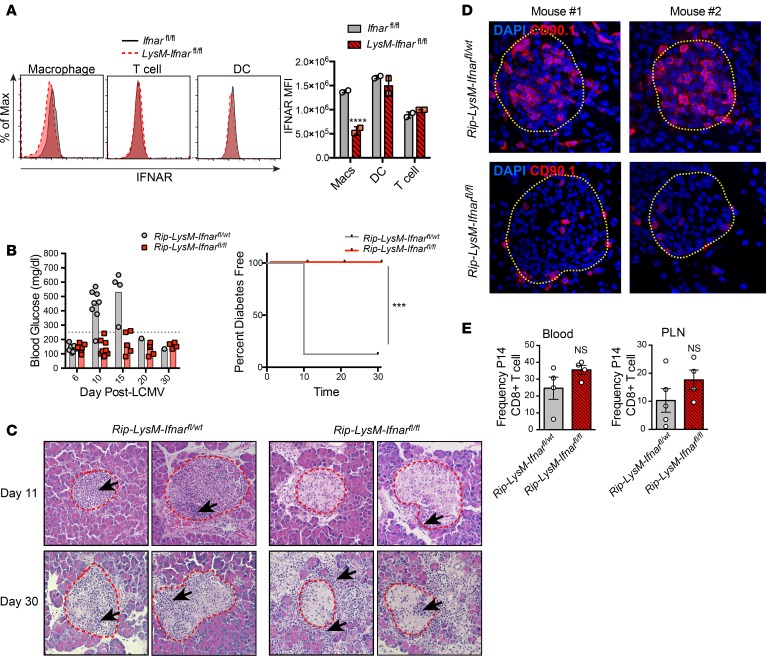
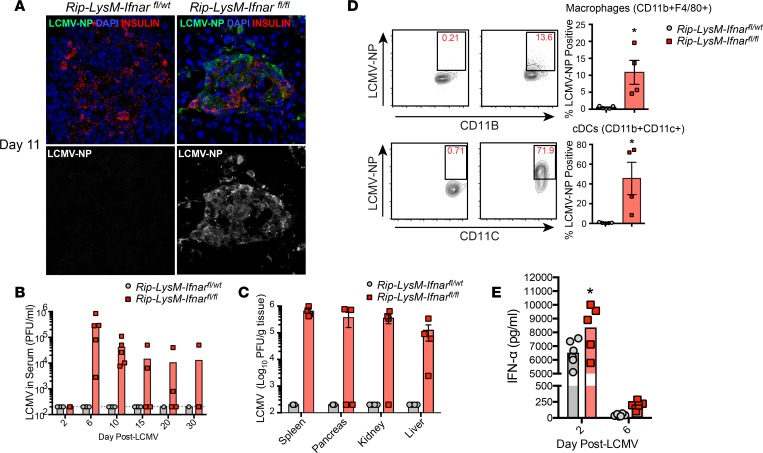
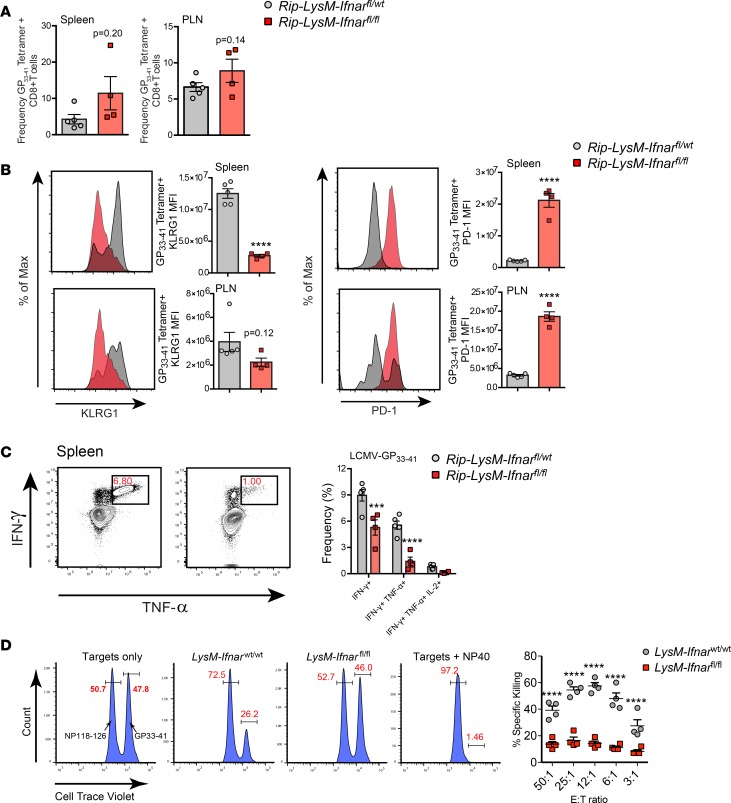
Here, we report a pathogenic role for type I IFN (IFN-I) signaling in macrophages, and not β cells in the islets, for the development of type 1 diabetes (T1D). Following lymphocytic choriomeningitis (LCMV) infection in the Rip-LCMV-GP T1D model, macrophages accumulated near islets and in close contact to islet-infiltrating GP-specific (autoimmune) CD8+ T cells. Depletion of macrophages with clodronate liposomes or genetic ablation of Ifnar in macrophages aborted T1D, despite proliferation of GP-specific (autoimmune) CD8+ T cells. Histopathologically, disrupted IFNα/β receptor (IFNAR) signaling in macrophages resulted in restriction of CD8+ T cells entering into the islets with significant lymphoid accumulation around the islet. Collectively, these results provide evidence that macrophages via IFN-I signaling, while not entering the islets, are directly involved in interacting, directing, or restricting trafficking of autoreactive-specific T cells into the islets as an important component in causing T1D.
Keywords: Autoimmunity; Cytokines; Diabetes; Immunology; Macrophages.
Conflict of interest statement
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
