

Single-cell RNA-seq denoising using a deep count autoencoder
- PMID: 30674886
- PMCID: PMC6344535
- DOI: 10.1038/s41467-018-07931-2
Single-cell RNA-seq denoising using a deep count autoencoder
Abstract
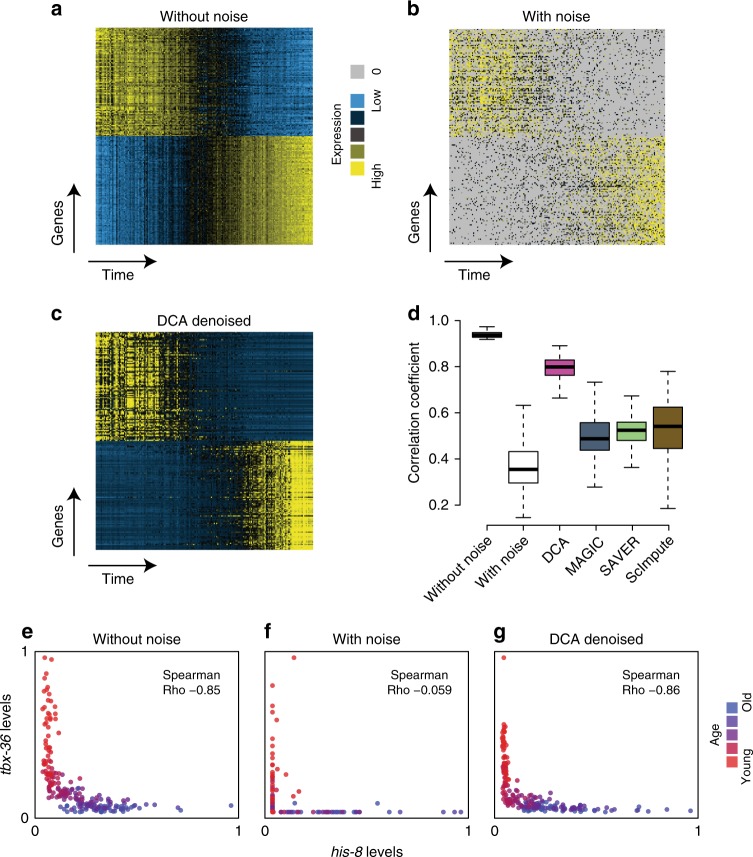
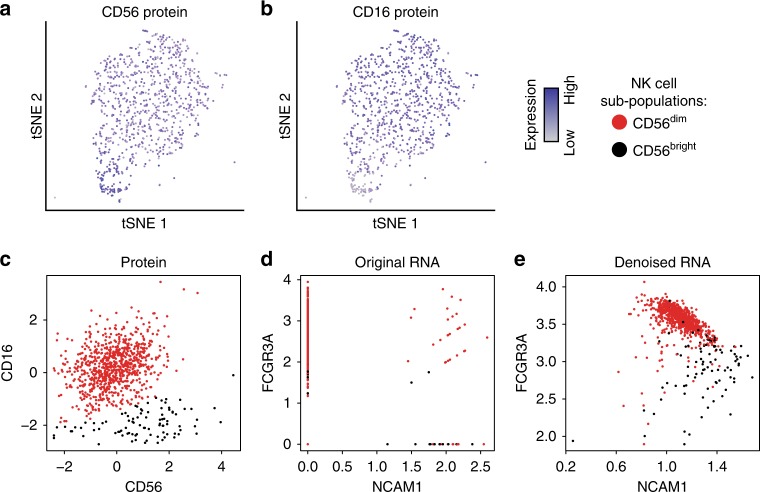
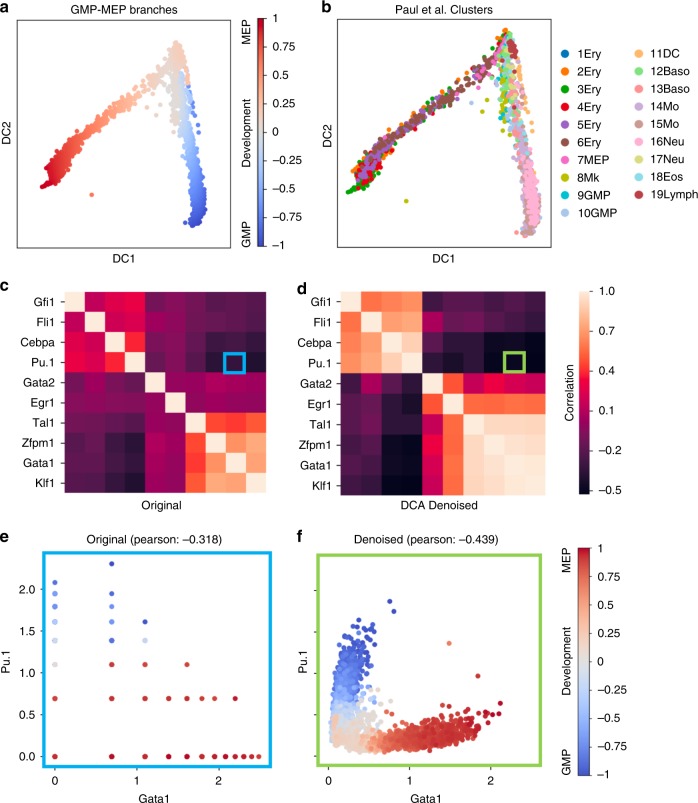
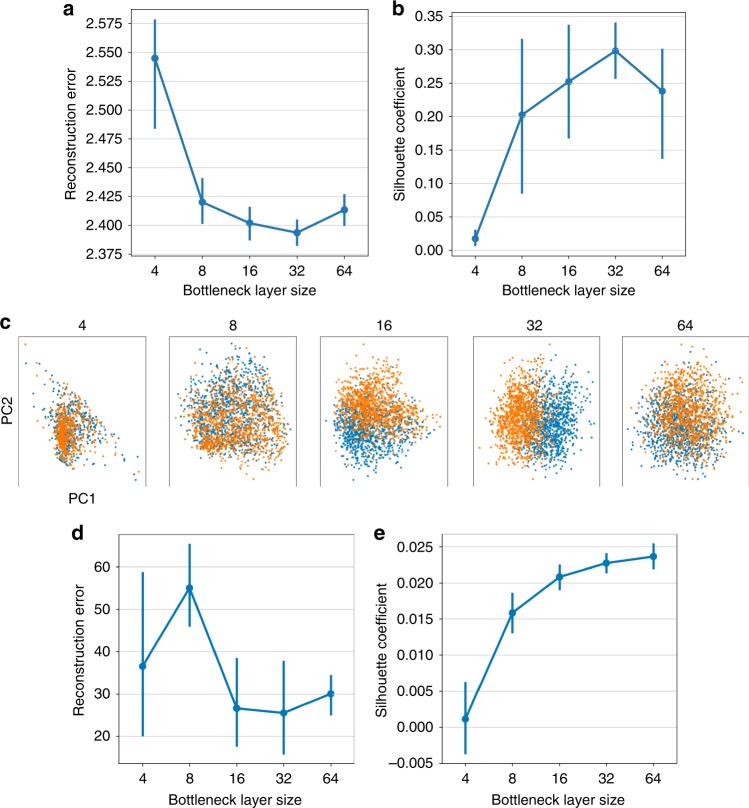
Single-cell RNA sequencing (scRNA-seq) has enabled researchers to study gene expression at a cellular resolution. However, noise due to amplification and dropout may obstruct analyses, so scalable denoising methods for increasingly large but sparse scRNA-seq data are needed. We propose a deep count autoencoder network (DCA) to denoise scRNA-seq datasets. DCA takes the count distribution, overdispersion and sparsity of the data into account using a negative binomial noise model with or without zero-inflation, and nonlinear gene-gene dependencies are captured. Our method scales linearly with the number of cells and can, therefore, be applied to datasets of millions of cells. We demonstrate that DCA denoising improves a diverse set of typical scRNA-seq data analyses using simulated and real datasets. DCA outperforms existing methods for data imputation in quality and speed, enhancing biological discovery.
Conflict of interest statement
The authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
