

Inhalation treatment of cystic fibrosis with lumacaftor and ivacaftor co-delivered by nanostructured lipid carriers
- PMID: 30677435
- PMCID: PMC6461390
- DOI: 10.1016/j.jconrel.2019.01.025
Inhalation treatment of cystic fibrosis with lumacaftor and ivacaftor co-delivered by nanostructured lipid carriers
Abstract
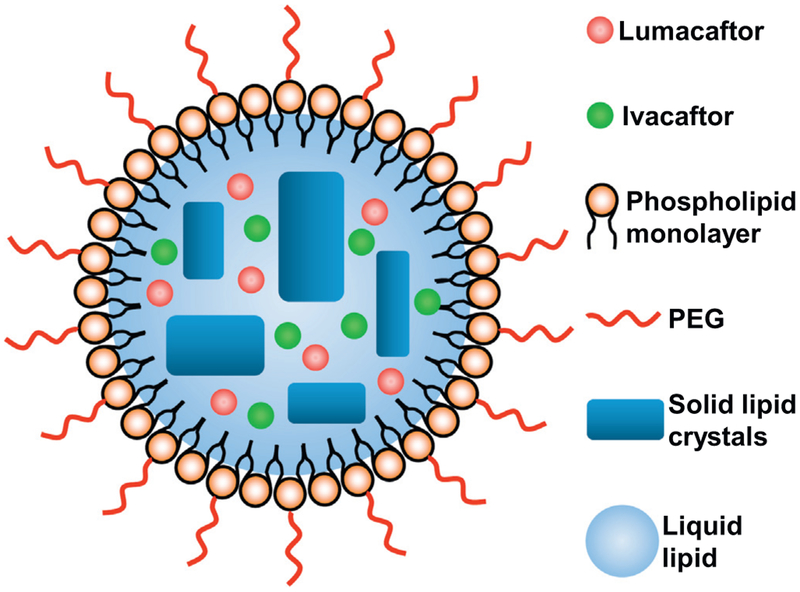
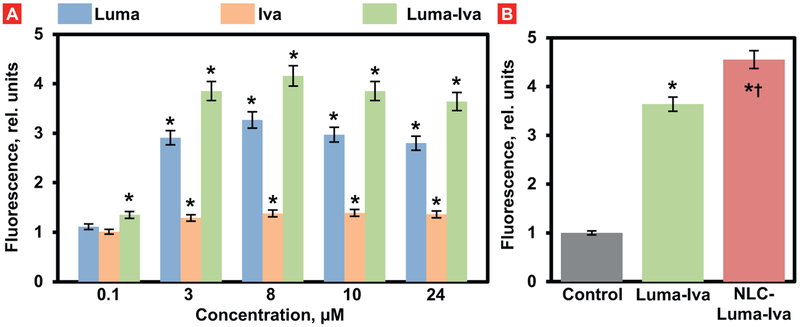
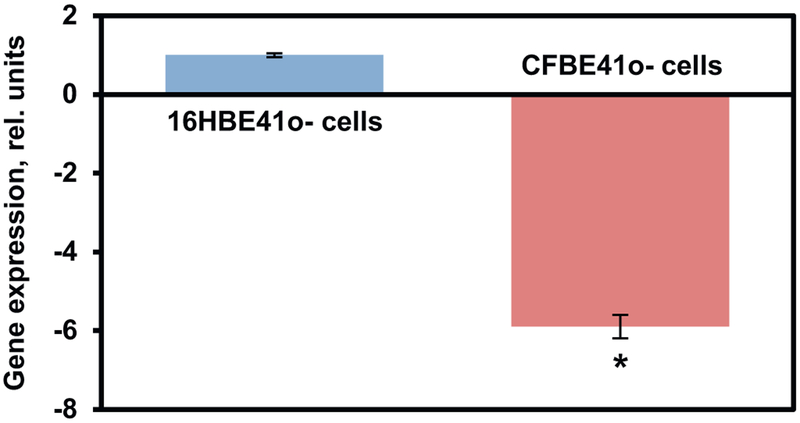
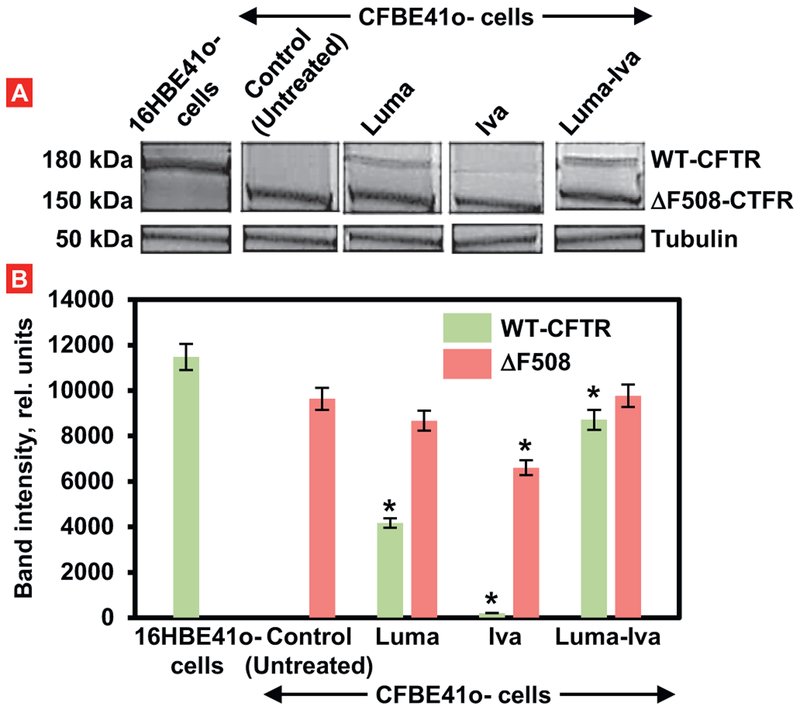
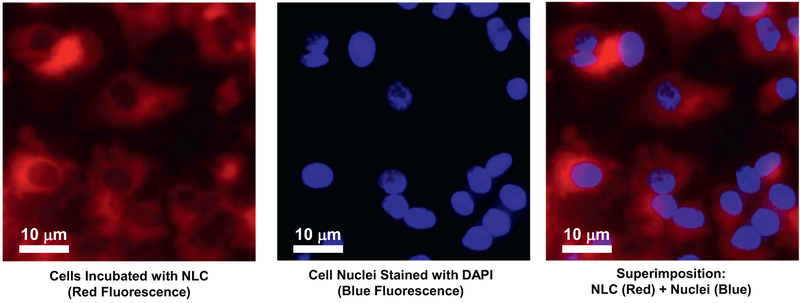
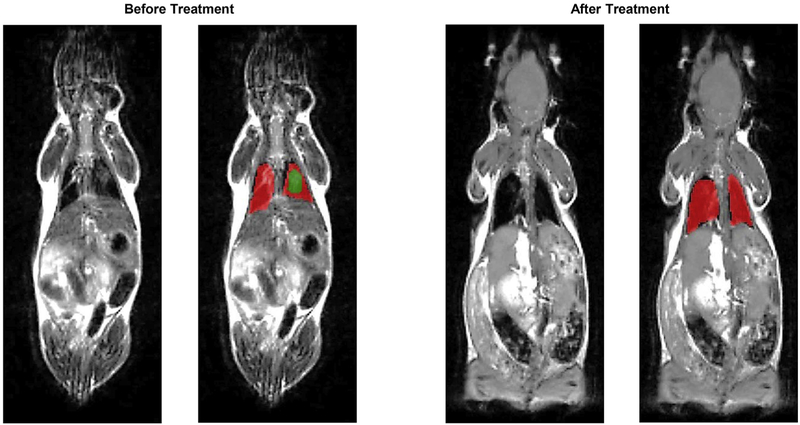
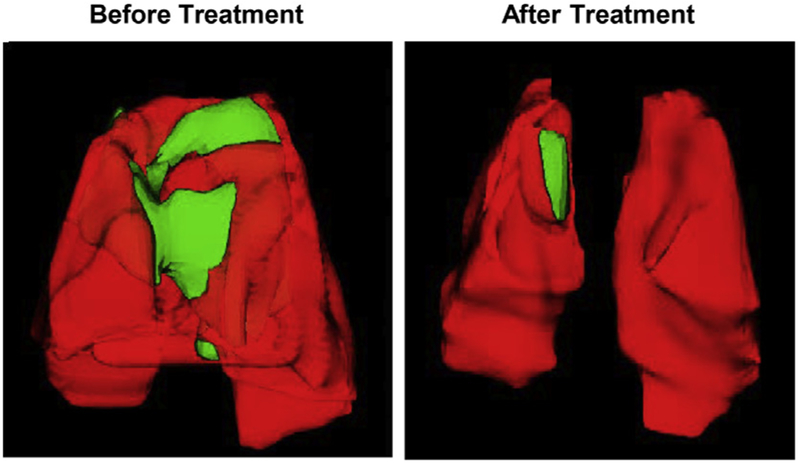
Cystic fibrosis (CF), a most deadly genetic disorder, is caused by mutations of CF transmembrane receptor (CFTR) - a chloride channel present at the surface of epithelial cells. In general, two steps have to be involved in treatment of the disease: correction of cellular defects and potentiation to further increase channel opening. Consequently, a combinatorial simultaneous treatment with two drugs with different mechanisms of action, lumacaftor and ivacaftor, has been recently proposed. While lumacaftor is used to correct p.Phe508del mutation (the loss of phenylalanine at position 508) and increase the amount of cell surface-localized CFTR protein, ivacaftor serves as a CFTR potentiator that increases the open probability of CFTR channels. Since the main organ that is affected by cystic fibrosis is the lung, the delivery of drugs directly to the lungs by inhalation has a potential to enhance the efficacy of the treatment of CF and limit adverse side effects upon healthy tissues and organs. Based on our extensive experience in inhalation delivering of drugs by different nanocarriers, we selected nanostructured lipid carriers (NLC) for the delivery both drugs directly to the lungs by inhalation and tested NLC loaded with drugs in vitro (normal and CF human bronchial epithelial cells) and in vivo (homozygote/homozygote bi-transgenic mice with CF). The results show that the designed NLCs demonstrated a high drug loading efficiency and were internalized in the cytoplasm of CF cells. It was found that NLC-loaded drugs were able to restore the expression and function of CFTR protein. As a result, the combination of lumacaftor and ivacaftor delivered by lipid nanoparticles directly into the lungs was highly effective in treating lung manifestations of cystic fibrosis.
Keywords: Cystic fibrosis transmembrane receptor (CFTR); Fibrosis; Imaging; Lipid nanoparticles; Pulmonary delivery; Transgenic mice.
Copyright © 2019 The Authors. Published by Elsevier B.V. All rights reserved.
Figures
References
-
- Welsh MJ, Smith AE, Molecular mechanisms of CFTR chloride channel dys-function in cystic fibrosis, Cell 73 (1993) 1251–1254. - PubMed
-
- Sorio C, Montresor A, Bolomini-Vittori M, Caldrer S, Rossi B, Dusi S, Angiari S, Johansson JE, Vezzalini M, Leal T, Calcaterra E, Assael BM, Melotti P, Laudanna C, Mutations of cystic fibrosis transmembrane conductance regulator gene cause a monocyte-selective adhesion deficiency, Am. J. Respir. Crit. Care Med 193 (2016) 1123–1133, 10.1164/rccm.201510-1922OC. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
