

Fast and flexible bacterial genomic epidemiology with PopPUNK
- PMID: 30679308
- PMCID: PMC6360808
- DOI: 10.1101/gr.241455.118
Fast and flexible bacterial genomic epidemiology with PopPUNK
Abstract
The routine use of genomics for disease surveillance provides the opportunity for high-resolution bacterial epidemiology. Current whole-genome clustering and multilocus typing approaches do not fully exploit core and accessory genomic variation, and they cannot both automatically identify, and subsequently expand, clusters of significantly similar isolates in large data sets spanning entire species. Here, we describe PopPUNK (
© 2019 Lees et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
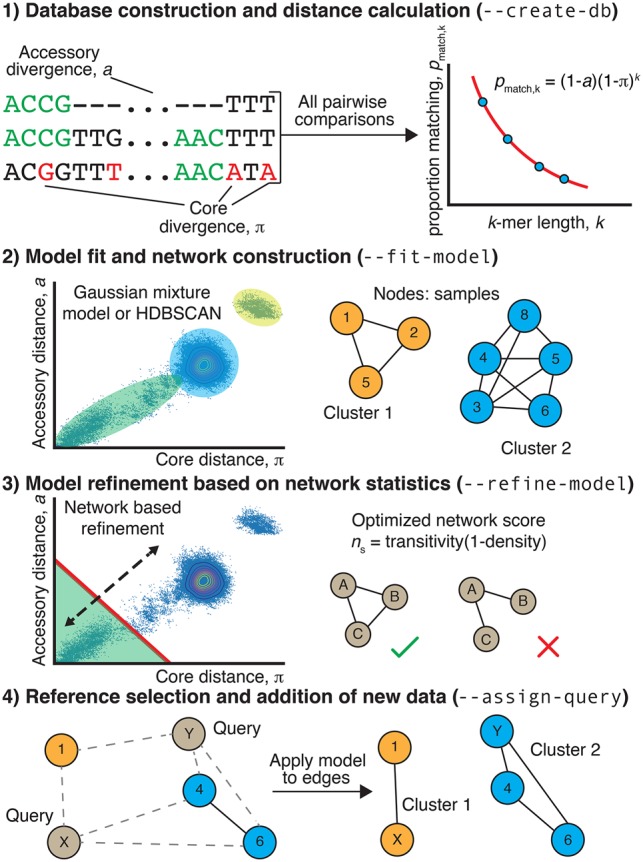
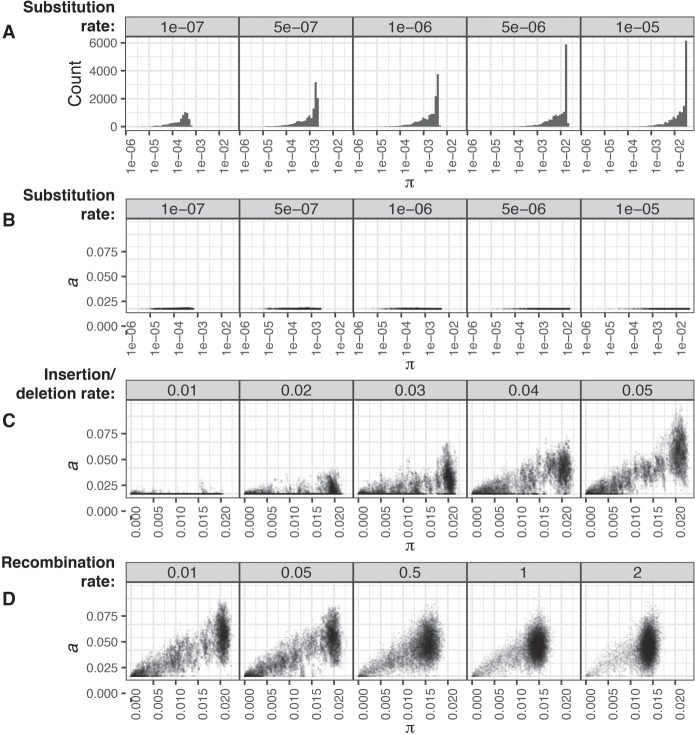
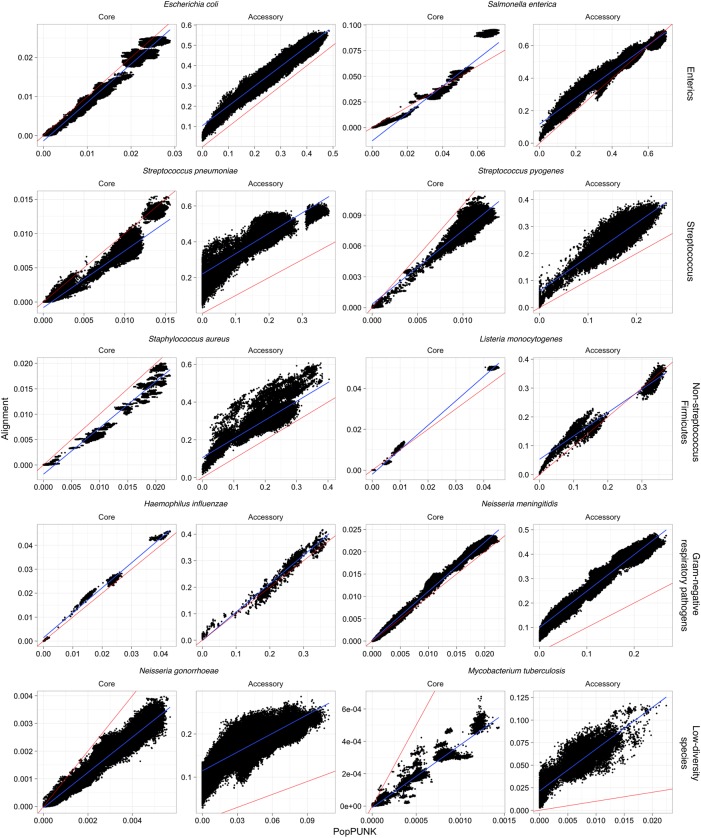
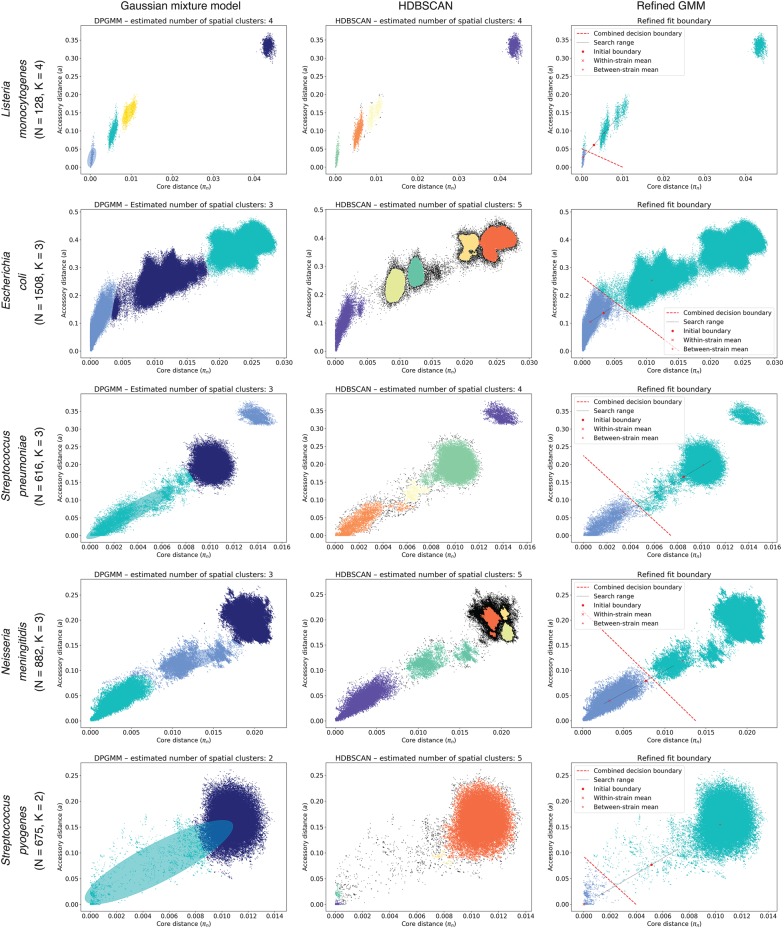
References
-
- Aanensen DM, Feil EJ, Holden MT, Dordel J, Yeats CA, Fedosejev A, Goater R, Castillo-Ramírez S, Corander J, Colijn C, et al. 2016. Whole-genome sequencing for routine pathogen surveillance in public health: a population snapshot of invasive Staphylococcus aureus in Europe. MBio 7: e00444-16 10.1128/mBio.00444-16 - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous