

EGF receptor and PKCδ kinase activate DNA damage-induced pro-survival and pro-apoptotic signaling via biphasic activation of ERK and MSK1 kinases
- PMID: 30679314
- PMCID: PMC6433046
- DOI: 10.1074/jbc.RA118.006944
EGF receptor and PKCδ kinase activate DNA damage-induced pro-survival and pro-apoptotic signaling via biphasic activation of ERK and MSK1 kinases
Abstract
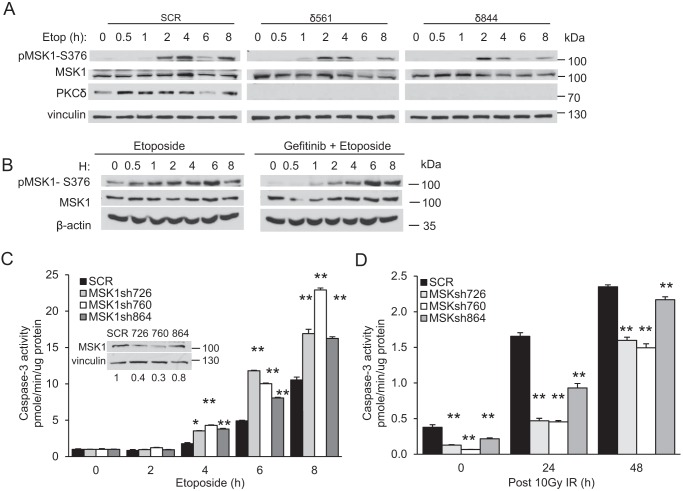
DNA damage-mediated activation of extracellular signal-regulated kinase (ERK) can regulate both cell survival and cell death. We show here that ERK activation in this context is biphasic and that early and late activation events are mediated by distinct upstream signals that drive cell survival and apoptosis, respectively. We identified the nuclear kinase mitogen-sensitive kinase 1 (MSK1) as a downstream target of both early and late ERK activation. We also observed that activation of ERK→MSK1 up to 4 h after DNA damage depends on epidermal growth factor receptor (EGFR), as EGFR or mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK)/ERK inhibitors or short hairpin RNA-mediated MSK1 depletion enhanced cell death. This prosurvival response was partially mediated through enhanced DNA repair, as EGFR or MEK/ERK inhibitors delayed DNA damage resolution. In contrast, the second phase of ERK→MSK1 activation drove apoptosis and required protein kinase Cδ (PKCδ) but not EGFR. Genetic disruption of PKCδ reduced ERK activation in an in vivo irradiation model, as did short hairpin RNA-mediated depletion of PKCδ in vitro In both models, PKCδ inhibition preferentially suppressed late activation of ERK. We have shown previously that nuclear localization of PKCδ is necessary and sufficient for apoptosis. Here we identified a nuclear PKCδ→ERK→MSK1 signaling module that regulates apoptosis. We also show that expression of nuclear PKCδ activates ERK and MSK1, that ERK activation is required for MSK1 activation, and that both ERK and MSK1 activation are required for apoptosis. Our findings suggest that location-specific activation by distinct upstream regulators may enable distinct functional outputs from common signaling pathways.
Keywords: DNA damage; apoptosis; cell signaling; epidermal growth factor receptor (EGFR); extracellular signal–regulated kinase (ERK); kinase cascade; mitogen-activated protein kinase (MAPK); mitogen-sensitive kinase (MSK); protein kinase C (PKC); signal transduction.
© 2019 Ohm et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Figures





Similar articles
-
Activation of ERK during DNA damage-induced apoptosis involves protein kinase Cdelta.Biochem Biophys Res Commun. 2005 Sep 9;334(4):1068-73. doi: 10.1016/j.bbrc.2005.06.199. Biochem Biophys Res Commun. 2005. PMID: 16039614
-
PKCδ regulates death receptor 5 expression induced by PS-341 through ATF4-ATF3/CHOP axis in human lung cancer cells.Mol Cancer Ther. 2012 Oct;11(10):2174-82. doi: 10.1158/1535-7163.MCT-12-0602. Epub 2012 Jul 30. Mol Cancer Ther. 2012. PMID: 22848091
-
Protein kinase Cdelta mediates neurogenic but not mitogenic activation of mitogen-activated protein kinase in neuronal cells.Mol Cell Biol. 1999 Jun;19(6):4209-18. doi: 10.1128/MCB.19.6.4209. Mol Cell Biol. 1999. PMID: 10330161 Free PMC article.
-
Protein kinase Cdelta and apoptosis.Biochem Soc Trans. 2007 Nov;35(Pt 5):1001-4. doi: 10.1042/BST0351001. Biochem Soc Trans. 2007. PMID: 17956263 Review.
-
PKCdelta signaling: mechanisms of DNA damage response and apoptosis.Cell Signal. 2007 May;19(5):892-901. doi: 10.1016/j.cellsig.2007.01.027. Epub 2007 Feb 7. Cell Signal. 2007. PMID: 17336499 Review.
Cited by
-
MAPK/ERK Pathway as a Central Regulator in Vertebrate Organ Regeneration.Int J Mol Sci. 2022 Jan 27;23(3):1464. doi: 10.3390/ijms23031464. Int J Mol Sci. 2022. PMID: 35163418 Free PMC article. Review.
-
Aging and Protein Kinases.Adv Exp Med Biol. 2021;1275:35-69. doi: 10.1007/978-3-030-49844-3_2. Adv Exp Med Biol. 2021. PMID: 33539011
-
KLF5-mediated Eppk1 expression promotes cell proliferation in cervical cancer via the p38 signaling pathway.BMC Cancer. 2021 Apr 8;21(1):377. doi: 10.1186/s12885-021-08040-y. BMC Cancer. 2021. PMID: 33827480 Free PMC article.
-
Targeting ERK-Hippo Interplay in Cancer Therapy.Int J Mol Sci. 2020 May 3;21(9):3236. doi: 10.3390/ijms21093236. Int J Mol Sci. 2020. PMID: 32375238 Free PMC article. Review.
-
Growth factor-induced activation of MSK2 leads to phosphorylation of H3K9me2S10 and corresponding changes in gene expression.Sci Adv. 2024 Mar 15;10(11):eadm9518. doi: 10.1126/sciadv.adm9518. Epub 2024 Mar 13. Sci Adv. 2024. PMID: 38478612 Free PMC article.
References
-
- Vissink A., Mitchell J. B., Baum B. J., Limesand K. H., Jensen S. B., Fox P. C., Elting L. S., Langendijk J. A., Coppes R. P., and Reyland M. E. (2010) Clinical management of salivary gland hypofunction and xerostomia in head-and-neck cancer patients: successes and barriers. Int. J. Radiat. Oncol. Biol. Phys. 78, 983–991 10.1016/j.ijrobp.2010.06.052 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
