

De novo variants in FBXO11 cause a syndromic form of intellectual disability with behavioral problems and dysmorphisms
- PMID: 30679813
- PMCID: PMC6462006
- DOI: 10.1038/s41431-018-0292-2
De novo variants in FBXO11 cause a syndromic form of intellectual disability with behavioral problems and dysmorphisms
Abstract
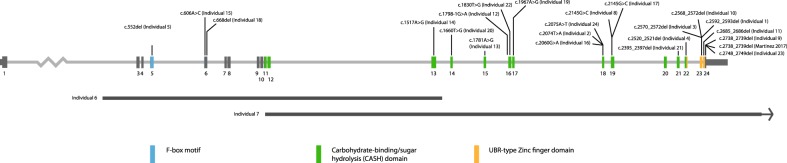
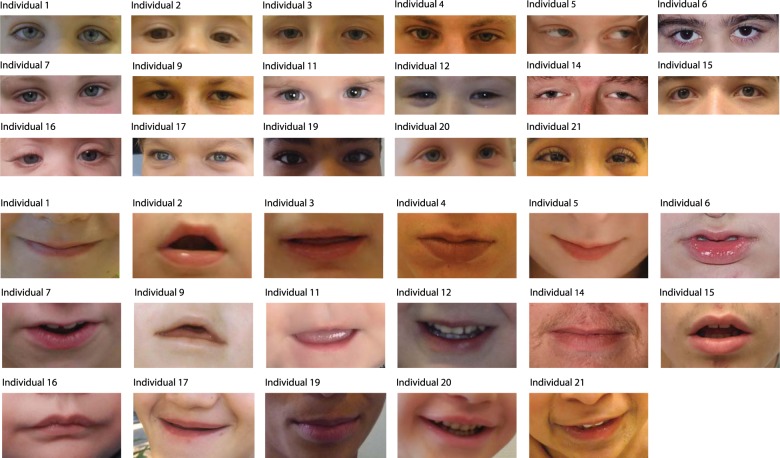
Determining pathogenicity of genomic variation identified by next-generation sequencing techniques can be supported by recurrent disruptive variants in the same gene in phenotypically similar individuals. However, interpretation of novel variants in a specific gene in individuals with mild-moderate intellectual disability (ID) without recognizable syndromic features can be challenging and reverse phenotyping is often required. We describe 24 individuals with a de novo disease-causing variant in, or partial deletion of, the F-box only protein 11 gene (FBXO11, also known as VIT1 and PRMT9). FBXO11 is part of the SCF (SKP1-cullin-F-box) complex, a multi-protein E3 ubiquitin-ligase complex catalyzing the ubiquitination of proteins destined for proteasomal degradation. Twenty-two variants were identified by next-generation sequencing, comprising 2 in-frame deletions, 11 missense variants, 1 canonical splice site variant, and 8 nonsense or frameshift variants leading to a truncated protein or degraded transcript. The remaining two variants were identified by array-comparative genomic hybridization and consisted of a partial deletion of FBXO11. All individuals had borderline to severe ID and behavioral problems (autism spectrum disorder, attention-deficit/hyperactivity disorder, anxiety, aggression) were observed in most of them. The most relevant common facial features included a thin upper lip and a broad prominent space between the paramedian peaks of the upper lip. Other features were hypotonia and hyperlaxity of the joints. We show that de novo variants in FBXO11 cause a syndromic form of ID. The current series show the power of reverse phenotyping in the interpretation of novel genetic variances in individuals who initially did not appear to have a clear recognizable phenotype.
Conflict of interest statement
M.J.G.S., K.G.M., and R.E.S. are employees of GeneDx Inc. H.R. is an employee of Impact Genetics Inc. The other authors declare that they have no conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
