

Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate-binding region
- PMID: 30682224
- PMCID: PMC6452443
- DOI: 10.1111/epi.14653
Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate-binding region
Abstract
Objective: To characterize the phenotypic spectrum associated with GNAO1 variants and establish genotype-protein structure-phenotype relationships.
Methods: We evaluated the phenotypes of 14 patients with GNAO1 variants, analyzed their variants for potential pathogenicity, and mapped them, along with those in the literature, on a three-dimensional structural protein model.
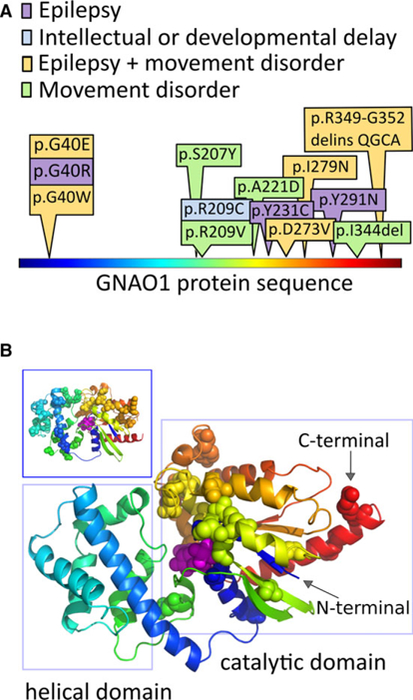
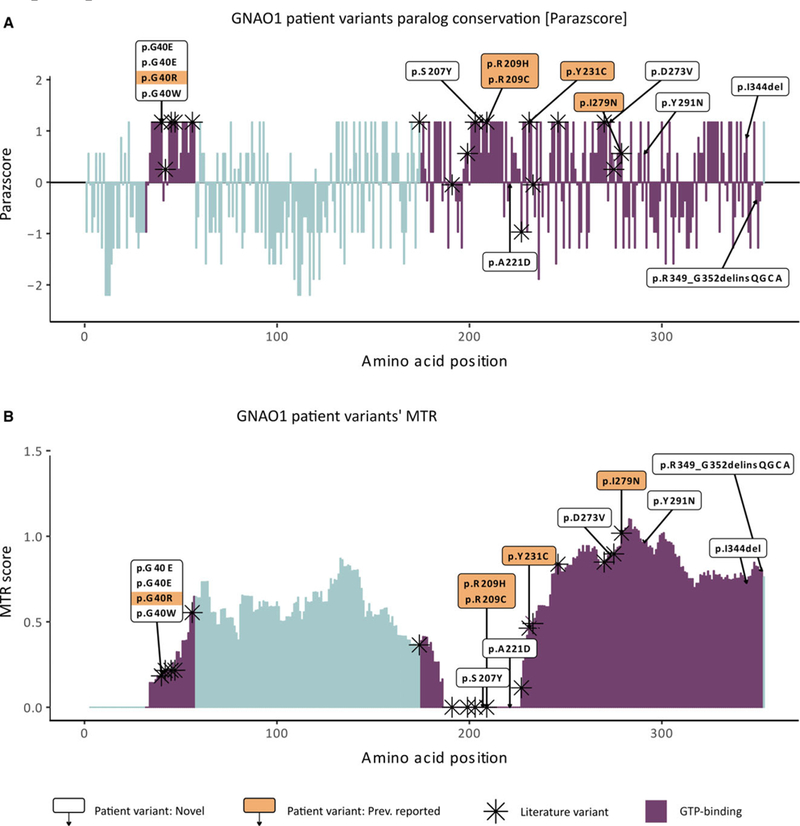
Results: The 14 patients in our cohort, including one sibling pair, had 13 distinct, heterozygous GNAO1 variants classified as pathogenic or likely pathogenic. We attributed the same variant in two siblings to parental mosaicism. Patients initially presented with seizures beginning in the first 3 months of life (8/14), developmental delay (4/14), hypotonia (1/14), or movement disorder (1/14). All patients had hypotonia and developmental delay ranging from mild to severe. Nine had epilepsy, and nine had movement disorders, including dystonia, ataxia, chorea, and dyskinesia. The 13 GNAO1 variants in our patients are predicted to result in amino acid substitutions or deletions in the GNAO1 guanosine triphosphate (GTP)-binding region, analogous to those in previous publications. Patients with variants affecting amino acids 207-221 had only movement disorder and hypotonia. Patients with variants affecting the C-terminal region had the mildest phenotypes.
Significance: GNAO1 encephalopathy most frequently presents with seizures beginning in the first 3 months of life. Concurrent movement disorders are also a prominent feature in the spectrum of GNAO1 encephalopathy. All variants affected the GTP-binding domain of GNAO1, highlighting the importance of this region for G-protein signaling and neurodevelopment.
Keywords: GNAO1; developmental and epileptic encephalopathy; mosaicism; movement disorders.
Wiley Periodicals, Inc. © 2019 International League Against Epilepsy.
Conflict of interest statement
DISCLOSURE
The authors have no conflicts of interest to report. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Figures
References
-
- Law CY, Chang ST, Cho SY, et al. Clinical whole‐exome equencing reveals a novel missense pathogenic variant of GNAO1 in a patient with infantile‐onset epilepsy. Clin Chim Acta 2015;451:292–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
