

A perilous path: the inborn errors of sphingolipid metabolism
- PMID: 30683667
- PMCID: PMC6399501
- DOI: 10.1194/jlr.S091827
A perilous path: the inborn errors of sphingolipid metabolism
Abstract
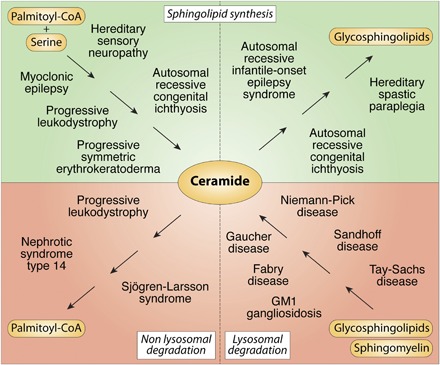
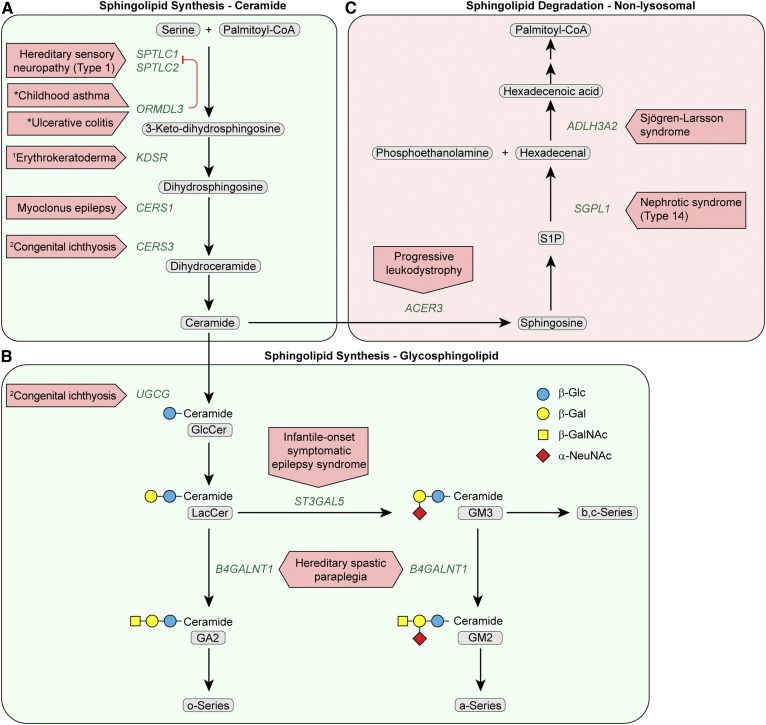
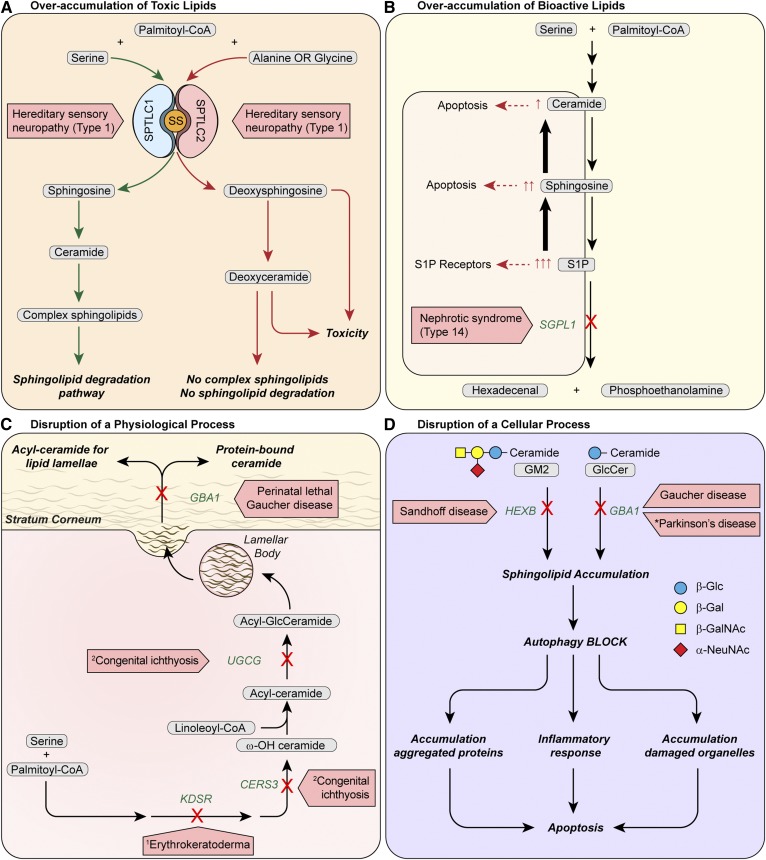
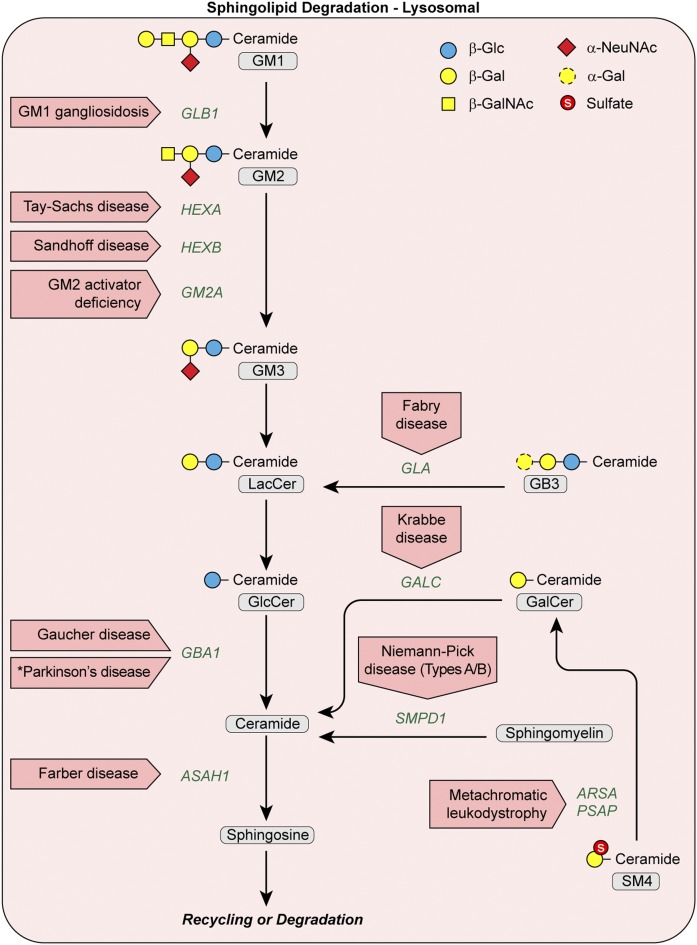
The sphingolipid (SL) metabolic pathway generates structurally diverse lipids that have roles as membrane constituents and as bioactive signaling molecules. The influence of the SL metabolic pathway in biology is pervasive; it exists in all mammalian cells and has roles in many cellular and physiological pathways. Human genetic diseases have long been recognized to be caused by mutations in the pathway, but until recently these mutational defects were only known to affect lysosomal SL degradation. Now, with a nearly complete delineation of the genes constituting the SL metabolic pathway, a growing number of additional genetic disorders caused by mutations in genes within other sectors of the pathway (de novo ceramide synthesis, glycosphingolipid synthesis, and nonlysosomal SL degradation) have been recognized. Although these inborn disorders of SL metabolism are clinically heterogeneous, some common pathogenic mechanisms, derived from the unique properties and functions of the SLs, underlie several of the diseases. These mechanisms include overaccumulation of toxic or bioactive lipids and the disruption of specific critical cellular and physiological processes. Many of these diseases also have commonalities in physiological systems affected, such as the nervous system and skin. While inborn disorders of SL metabolism are rare, gene variants in the pathway have been linked to increased susceptibility to Parkinson's disease and childhood asthma, implying that the SL metabolic pathway may have a role in these disorders. A more complete understanding of the inborn errors of SL metabolism promises new insights into the convergence of their pathogenesis with those of common human diseases.
Keywords: bioactive lipids; ceramides; gangliosides; genetics; glycosphingolipids; metabolic disease; rare disease; sphingolipids; storage diseases.
Copyright © 2019 Dunn et al. Published by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that there are no conflicts of interest with the contents of this article.
Figures
References
-
- Rabionet M., Gorgas K., and Sandhoff R.. 2014. Ceramide synthesis in the epidermis. Biochim. Biophys. Acta. 1841: 422–434. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
