

Congenital erythropoietic porphyria: Recent advances
- PMID: 30685241
- PMCID: PMC6597325
- DOI: 10.1016/j.ymgme.2018.12.008
Congenital erythropoietic porphyria: Recent advances
Abstract
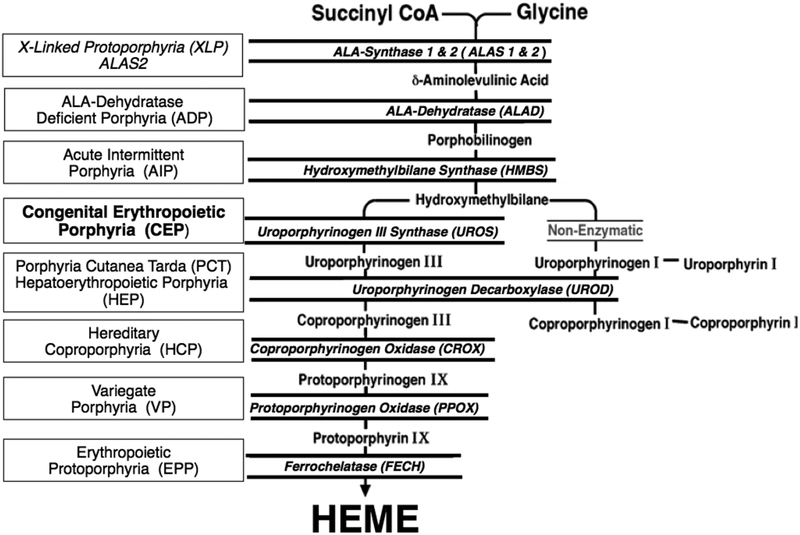
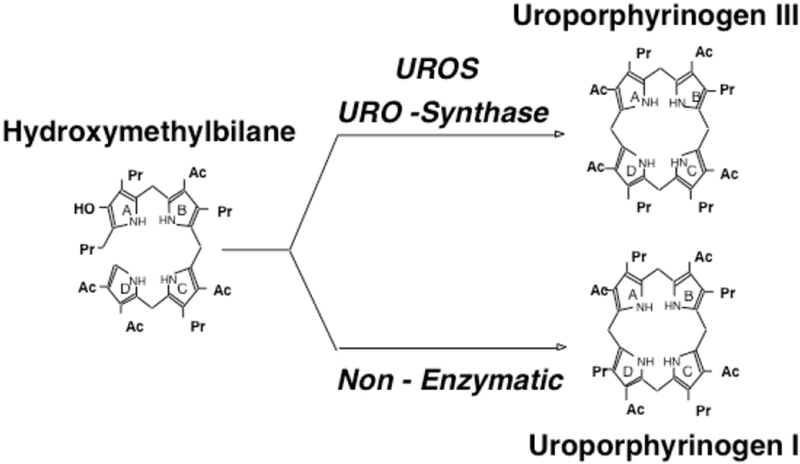
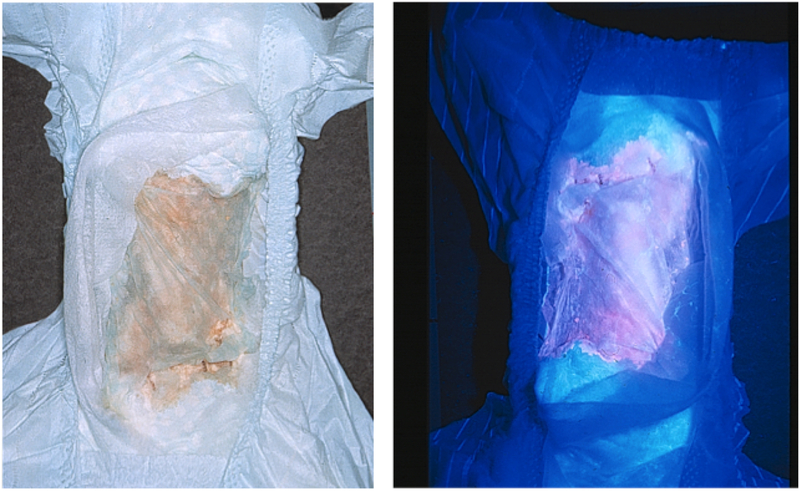
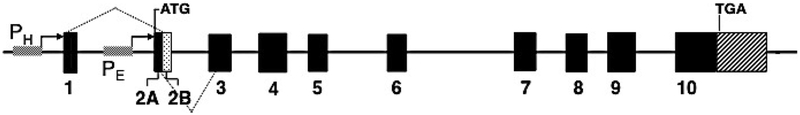
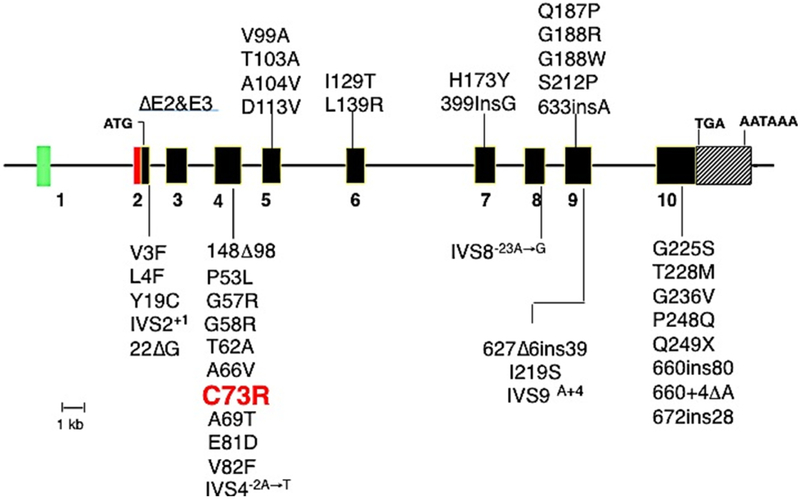
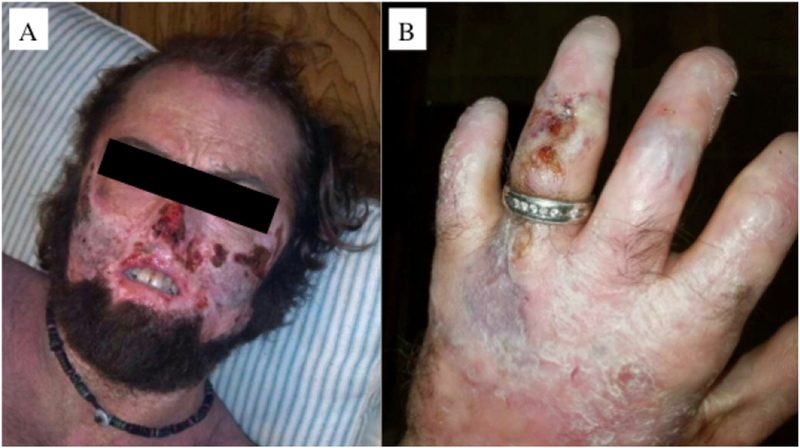
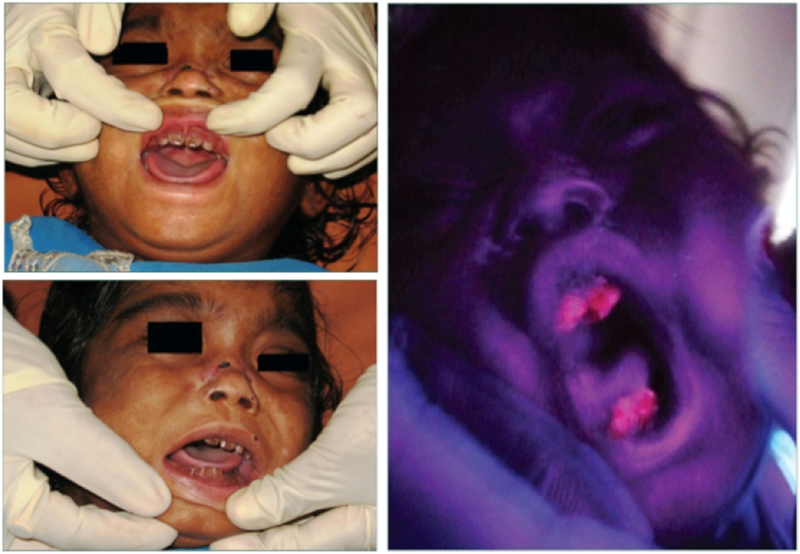
Congenital erythropoietic porphyria (CEP) is a rare autosomal recessive disorder characterized by photosensitivity and by hematologic abnormalities in affected individuals. CEP is caused by mutations in the uroporphyrinogen synthase (UROS) gene. In three reported cases, CEP has been associated with a specific X-linked GATA1 mutation. Disease-causing mutations in either gene result in absent or markedly reduced UROS enzymatic activity. This in turn leads to the accumulation of the non-physiologic and photoreactive porphyrinogens, uroporphyrinogen I and coproporphyrinogen I, which damage erythrocytes and elicit a phototoxic reaction upon light exposure. The clinical spectrum of CEP depends on the level of residual UROS activity, which is determined by the underlying pathogenic loss-of-function UROS mutations. Disease severity ranges from non-immune hydrops fetalis in utero to late-onset disease with only mild cutaneous involvement. The clinical characteristics of CEP include exquisite photosensitivity to visible light resulting in bullous vesicular lesions which, when infected lead to progressive photomutilation of sun-exposed areas such as the face and hands. In addition, patients have erythrodontia (brownish discoloration of teeth) and can develop corneal scarring. Chronic transfusion-dependent hemolytic anemia is common and leads to bone marrow hyperplasia, which further increases porphyrin production. Management of CEP consists of strict avoidance of exposure to visible light with sun-protective clothing, sunglasses, and car and home window filters. Adequate care of ruptured vesicles and use of topical antibiotics is indicated to prevent superinfections and osteolysis. In patients with symptomatic hemolytic anemia, frequent erythrocyte cell transfusions may be necessary to suppress hematopoiesis and decrease marrow production of the phototoxic porphyrins. In severe transfection-dependent cases, bone marrow or hematopoietic stem cell transplantation has been performed, which is curative. Therapeutic approaches including gene therapy, proteasome inhibition, and pharmacologic chaperones are under investigation.
Keywords: Cutaneous lesions, Heme biosynthetic pathway; Hemolysis; Phototoxicity; Porphyria.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of Interest:
The authors report no relevant conflicts
Figures
References
-
- Anderson KE, Sassa S, Bishop DF, Desnick RJ, Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias In: Metabolic The and Molecular Bases of Inherited Disease, 8th (ed), Scriver CR, Beaudet AL, Sly WS and Valle D, (eds), New York (NY), McGraw-Hill, (2014) 2961–3062. http://ommbid.mhmedical.com/content.aspx?bookid=971&Sectionid=62638866.
-
- Puy H, Gouya L, Deybach JC, Porphyrias. Lancet 375 (2010) 924–937. - PubMed
-
- Bickers DR, Frank J, The Porphyrias. In: Fitzpatrick’s Dermatology in General Medicine, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K (eds), 8th (ed), McGraw-Hill, New York (NY), Chapter 132, (2012) 1679 https://accessmedicine.mhmedical.com/Content.aspx?bookId=392§ionId=4....
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
