

Gene Augmentation and Readthrough Rescue Channelopathy in an iPSC-RPE Model of Congenital Blindness
- PMID: 30686507
- PMCID: PMC6369573
- DOI: 10.1016/j.ajhg.2018.12.019
Gene Augmentation and Readthrough Rescue Channelopathy in an iPSC-RPE Model of Congenital Blindness
Abstract
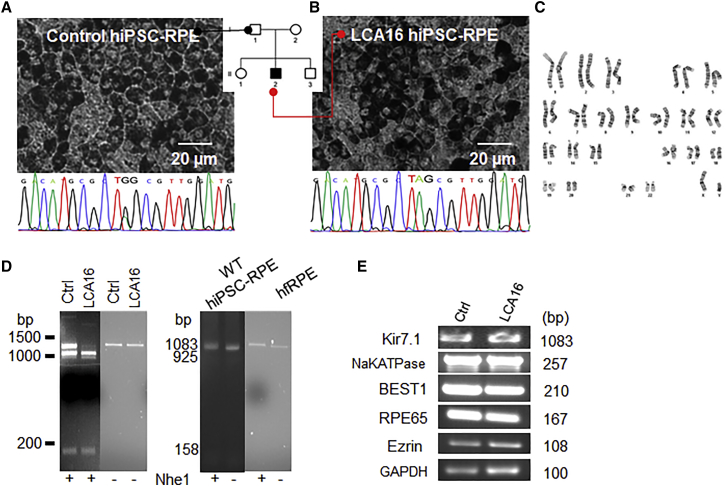
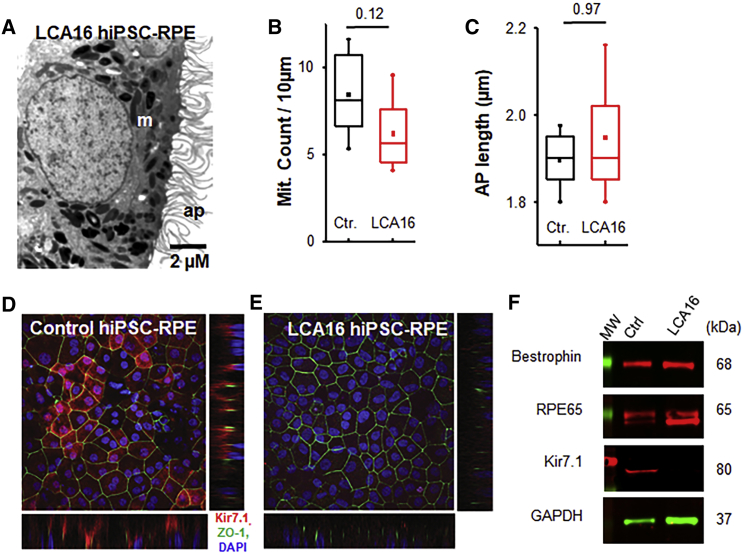
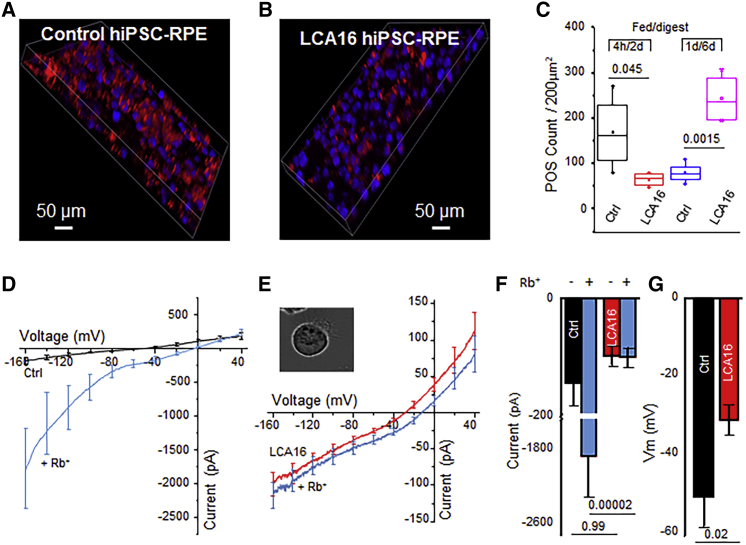
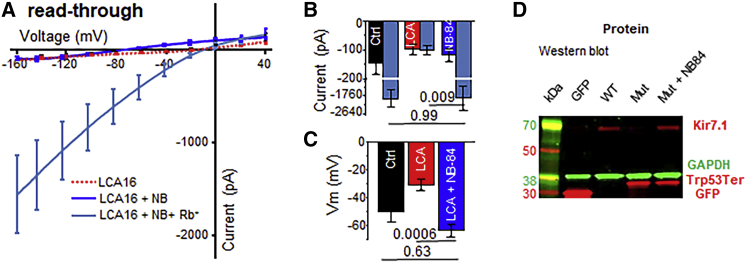
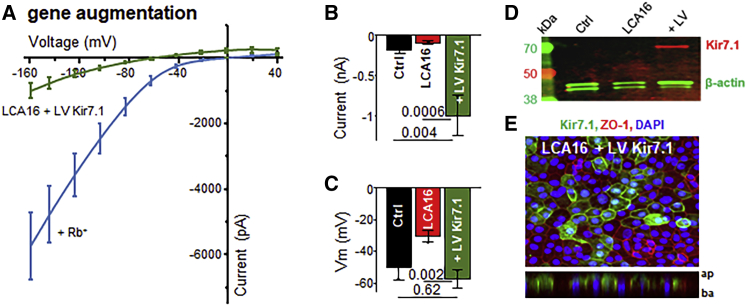
Pathogenic variants of the KCNJ13 gene are known to cause Leber congenital amaurosis (LCA16), an inherited pediatric blindness. KCNJ13 encodes the Kir7.1 subunit that acts as a tetrameric, inwardly rectifying potassium ion channel in the retinal pigment epithelium (RPE) to maintain ionic homeostasis and allow photoreceptors to encode visual information. We sought to determine whether genetic approaches might be effective in treating blindness arising from pathogenic variants in KCNJ13. We derived human induced pluripotent stem cell (hiPSC)-RPE cells from an individual carrying a homozygous c.158G>A (p.Trp53∗) pathogenic variant of KCNJ13. We performed biochemical and electrophysiology assays to confirm Kir7.1 function. We tested both small-molecule readthrough drug and gene-therapy approaches for this "disease-in-a-dish" approach. We found that the LCA16 hiPSC-RPE cells had normal morphology but did not express a functional Kir7.1 channel and were unable to demonstrate normal physiology. After readthrough drug treatment, the LCA16 hiPSC cells were hyperpolarized by 30 mV, and the Kir7.1 current was restored. Similarly, we rescued Kir7.1 channel function after lentiviral gene delivery to the hiPSC-RPE cells. In both approaches, Kir7.1 was expressed normally, and there was restoration of membrane potential and the Kir7.1 current. Loss-of-function variants of Kir7.1 are one cause of LCA. Using either readthrough therapy or gene augmentation, we rescued Kir7.1 channel function in iPSC-RPE cells derived from an affected individual. This supports the development of precision-medicine approaches for the treatment of clinical LCA16.
Keywords: KCNJ13; Kir7.1; Leber congenital amaurosis (LCA); blindness; gene-therapy; genetic disorders; induced pluripotent stem cells (iPSC); ion-channels; loss-of-function; retinal pigment epithelium (RPE); translational readthrough.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Pattnaik B.R., Shahi P.K., Marino M.J., Liu X., York N., Brar S., Chiang J., Pillers D.A., Traboulsi E.I. A novel KCNJ13 nonsense mutation and loss of Kir7.1 channel function causes Leber congenital amaurosis (LCA16) Hum. Mutat. 2015;36:720–727. - PubMed
-
- Perez-Roustit S., Marquette V., Bocquet B., Kaplan J., Perrault I., Meunier I., Hamel C.P. Leber congenital amaurosis with large retinal pigment clumps caused by compound heterozygous mutations in Kcnj13. Retin. Cases Brief Rep. 2017;11:221–226. - PubMed
-
- Khan A.O., Bergmann C., Neuhaus C., Bolz H.J. A distinct vitreo-retinal dystrophy with early-onset cataract from recessive KCNJ13 mutations. Ophthalmic Genet. 2015;36:79–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
