

Evidence for a recombinant origin of HIV-1 Group M from genomic variation
- PMID: 30687518
- PMCID: PMC6342232
- DOI: 10.1093/ve/vey039
Evidence for a recombinant origin of HIV-1 Group M from genomic variation
Abstract
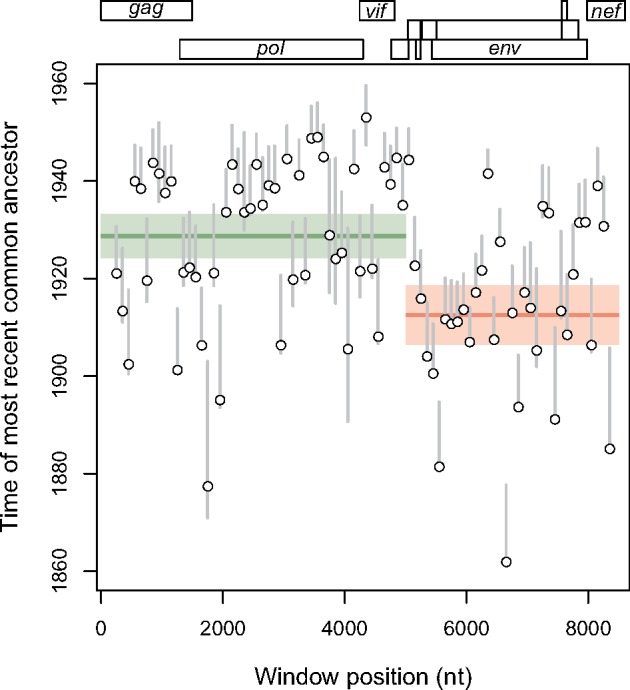
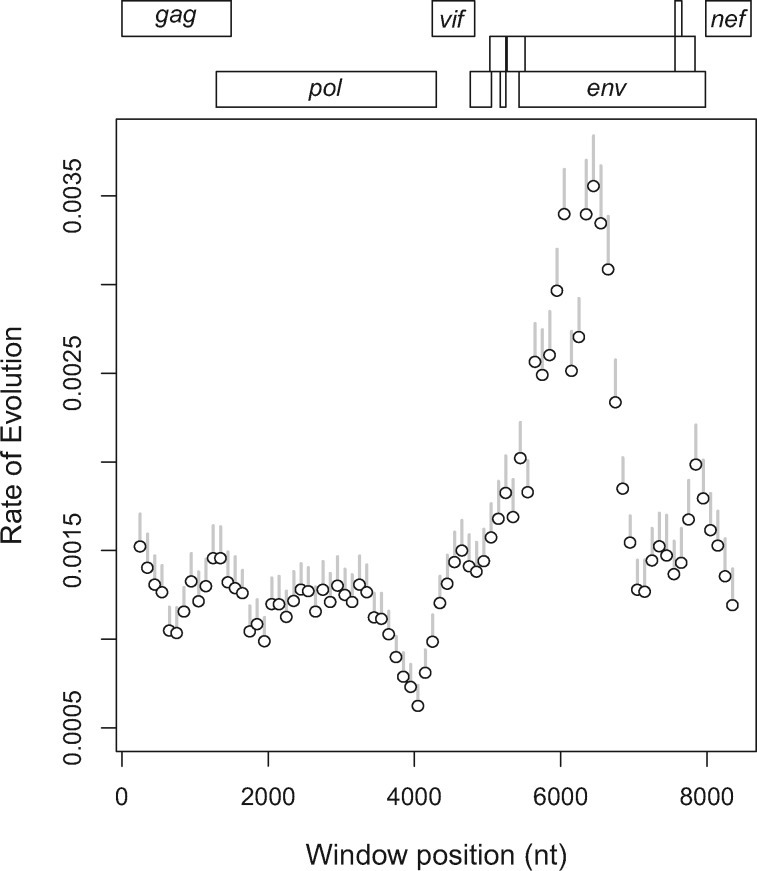
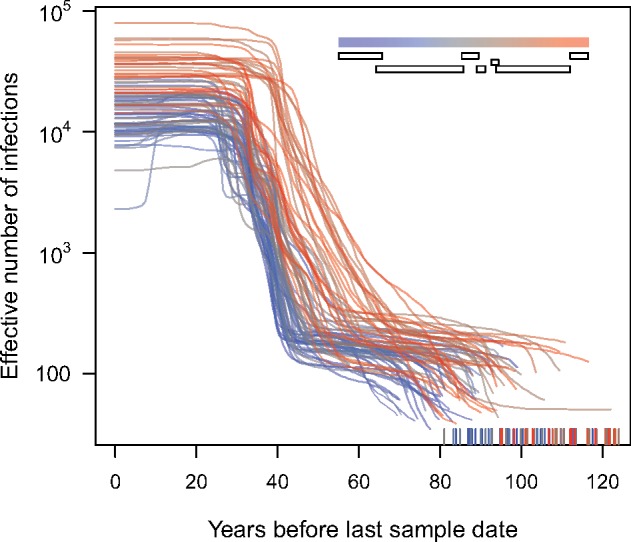
Reconstructing the early dynamics of the HIV-1 pandemic can provide crucial insights into the socioeconomic drivers of emerging infectious diseases in human populations, including the roles of urbanization and transportation networks. Current evidence indicates that the global pandemic comprising almost entirely of HIV-1/M originated around the 1920s in central Africa. However, these estimates are based on molecular clock estimates that are assumed to apply uniformly across the virus genome. There is growing evidence that recombination has played a significant role in the early history of the HIV-1 pandemic, such that different regions of the HIV-1 genome have different evolutionary histories. In this study, we have conducted a dated-tip analysis of all near full-length HIV-1/M genome sequences that were published in the GenBank database. We used a sliding window approach similar to the 'bootscanning' method for detecting breakpoints in inter-subtype recombinant sequences. We found evidence of substantial variation in estimated root dates among windows, with an estimated mean time to the most recent common ancestor of 1922. Estimates were significantly autocorrelated, which was more consistent with an early recombination event than with stochastic error variation in phylogenetic reconstruction and dating analyses. A piecewise regression analysis supported the existence of at least one recombination breakpoint in the HIV-1/M genome with interval-specific means around 1929 and 1913, respectively. This analysis demonstrates that a sliding window approach can accommodate early recombination events outside the established nomenclature of HIV-1/M subtypes, although it is difficult to incorporate the earliest available samples due to their limited genome coverage.
Keywords: HIV-1; molecular clock; network clustering; phylogenetics; recombination; subtype diversity.
Figures

References
-
- Anderson T. K. et al. (2012) ‘Ranking Viruses: Measures of Positional Importance within Networks Define Core Viruses for Rational Polyvalent Vaccine Development’, Bioinformatics, 28: 1624–32. - PubMed
LinkOut - more resources
Full Text Sources
