

Epidermolysis Bullosa Acquisita: The 2019 Update
- PMID: 30687710
- PMCID: PMC6335340
- DOI: 10.3389/fmed.2018.00362
Epidermolysis Bullosa Acquisita: The 2019 Update
Abstract
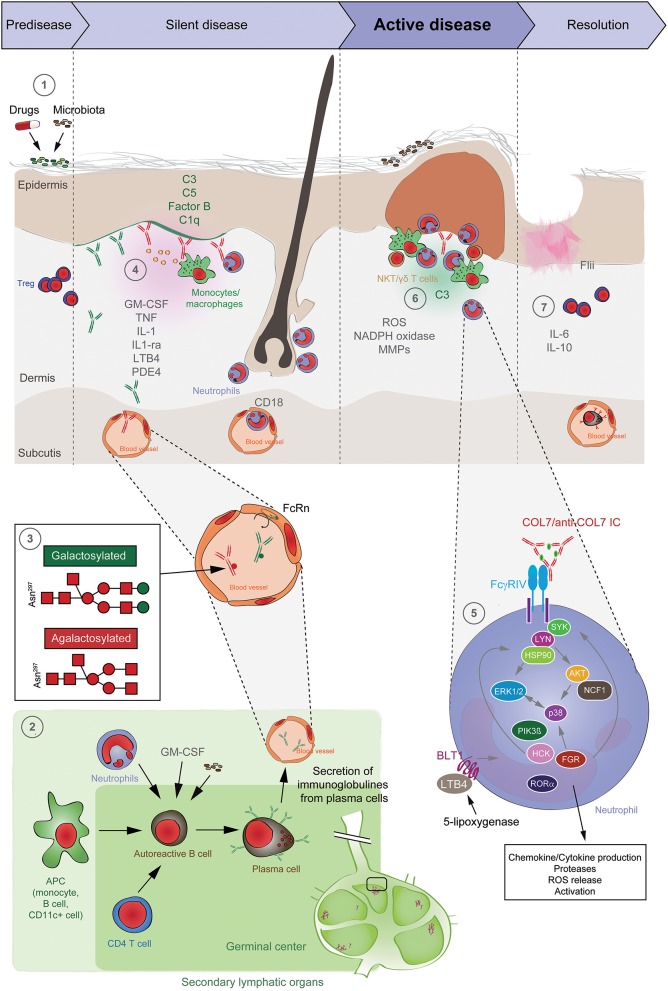
Epidermolysis bullosa acquisita (EBA) is an orphan autoimmune disease. Patients with EBA suffer from chronic inflammation as well as blistering and scarring of the skin and mucous membranes. Current treatment options rely on non-specific immunosuppression, which in many cases, does not lead to a remission of treatment. Hence, novel treatment options are urgently needed for the care of EBA patients. During the past decade, decisive clinical observations, and frequent use of pre-clinical model systems have tremendously increased our understanding of EBA pathogenesis. Herein, we review all of the aspects of EBA, starting with a detailed description of epidemiology, clinical presentation, diagnosis, and current treatment options. Of note, pattern analysis via direct immunofluorescence microscopy of a perilesional skin lesion and novel serological test systems have significantly facilitated diagnosis of the disease. Next, a state-of the art review of the current understanding of EBA pathogenesis, emerging treatments and future perspectives is provided. Based on pre-clinical model systems, cytokines and kinases are among the most promising therapeutic targets, whereas high doses of IgG (IVIG) and the anti-CD20 antibody rituximab are among the most promising "established" EBA therapeutics. We also aim to raise awareness of EBA, as well as initiate basic and clinical research in this field, to further improve the already improved but still unsatisfactory conditions for those diagnosed with this condition.
Keywords: animal models; diagnosis; epidermolysis bullosa acquisita; pathogenesis; treatment.
Figures
Similar articles
-
Meta-analysis of the clinical and immunopathological characteristics and treatment outcomes in epidermolysis bullosa acquisita patients.Orphanet J Rare Dis. 2018 Sep 4;13(1):153. doi: 10.1186/s13023-018-0896-1. Orphanet J Rare Dis. 2018. PMID: 30180870 Free PMC article.
-
Clinical features and diagnosis of epidermolysis bullosa acquisita.Expert Rev Clin Immunol. 2017 Feb;13(2):157-169. doi: 10.1080/1744666X.2016.1221343. Epub 2016 Sep 8. Expert Rev Clin Immunol. 2017. PMID: 27580464 Review.
-
Pathogenesis of epidermolysis bullosa acquisita.Dermatol Clin. 2011 Jul;29(3):493-501, xi. doi: 10.1016/j.det.2011.03.003. Dermatol Clin. 2011. PMID: 21605817 Review.
-
Model systems duplicating epidermolysis bullosa acquisita: a methodological review.Autoimmunity. 2012 Feb;45(1):102-10. doi: 10.3109/08916934.2011.606451. Epub 2011 Sep 19. Autoimmunity. 2012. PMID: 21923614 Review.
-
Epidermolysis bullosa acquisita: A comprehensive review.Autoimmun Rev. 2019 Aug;18(8):786-795. doi: 10.1016/j.autrev.2019.06.007. Epub 2019 Jun 7. Autoimmun Rev. 2019. PMID: 31181325 Review.
Cited by
-
There Is Strength in Numbers: Quantitation of Fc Gamma Receptors on Murine Tissue-Resident Macrophages.Int J Mol Sci. 2021 Nov 10;22(22):12172. doi: 10.3390/ijms222212172. Int J Mol Sci. 2021. PMID: 34830050 Free PMC article.
-
Drug Development in Pemphigoid Diseases.Acta Derm Venereol. 2020 Feb 12;100(5):adv00055. doi: 10.2340/00015555-3400. Acta Derm Venereol. 2020. PMID: 32039458 Free PMC article. Review.
-
Epidermolysis bullosa acquisita treated with rituximab.BMJ Case Rep. 2021 Jul 13;14(7):e243432. doi: 10.1136/bcr-2021-243432. BMJ Case Rep. 2021. PMID: 34257124 Free PMC article. No abstract available.
-
Off-Label Uses of Rituximab in Dermatology.Curr Dermatol Rep. 2022;11(4):209-220. doi: 10.1007/s13671-022-00375-4. Epub 2022 Oct 6. Curr Dermatol Rep. 2022. PMID: 36217351 Free PMC article. Review.
-
Genetics and Omics Analysis of Autoimmune Skin Blistering Diseases.Front Immunol. 2019 Oct 15;10:2327. doi: 10.3389/fimmu.2019.02327. eCollection 2019. Front Immunol. 2019. PMID: 31749790 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous