

Predicting protein-ligand binding modes for CELPP and GC3: workflows and insight
- PMID: 30689079
- PMCID: PMC6494980
- DOI: 10.1007/s10822-019-00185-0
Predicting protein-ligand binding modes for CELPP and GC3: workflows and insight
Abstract
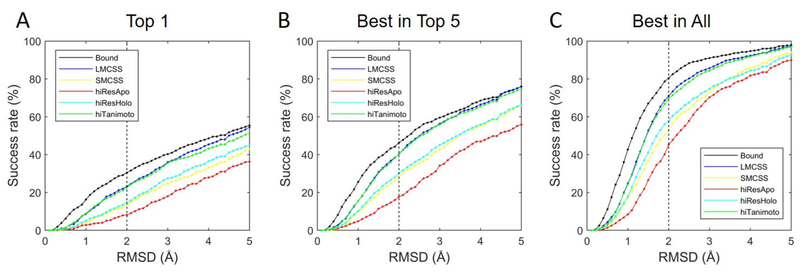
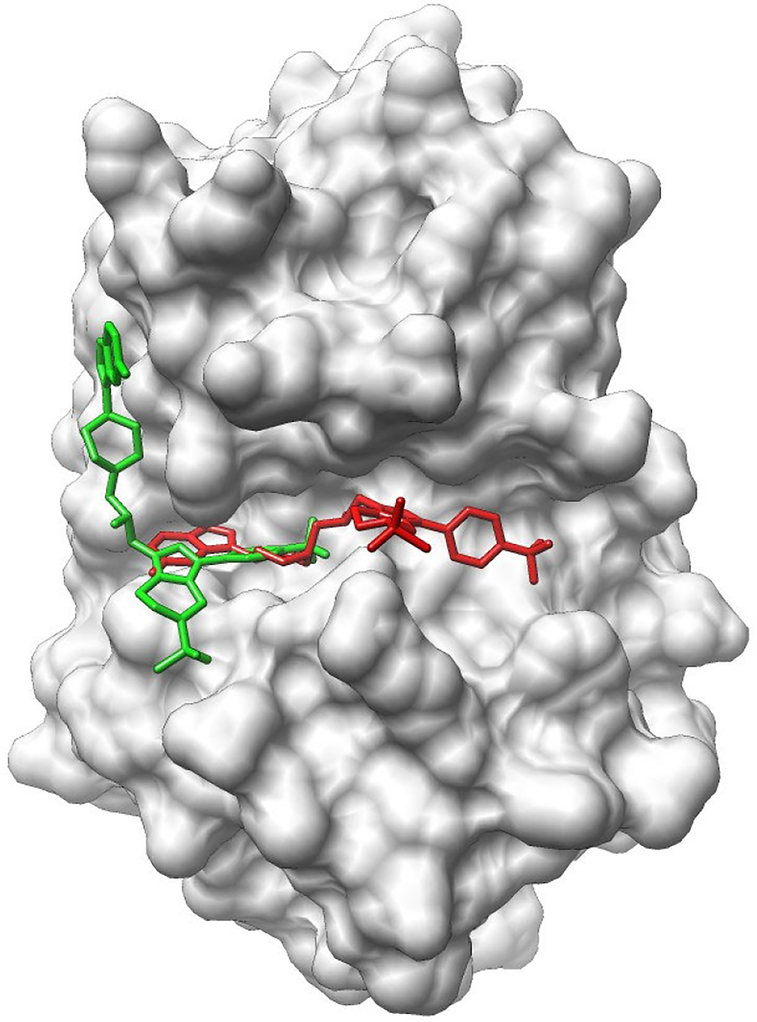
Drug Design Data Resource (D3R) continues to release valuable benchmarking datasets to promote improvement and development of computational methods for new drug discovery. We have developed several methods for protein-ligand binding mode prediction during the participation in the D3R challenges. In the present study, these methods were integrated, automated, and systematically tested using the large-scale data from Continuous Evaluation of Ligand Pose Prediction (CELPP) and a subset of Grand challenge 3 (GC3). The results show that current molecular docking methods benefit from the increasing number of protein-ligand complex structures deposited in Protein Data Bank. Using an appropriate protein structure for docking significantly improves the success rate of the binding mode prediction. The results of our template-based method and docking method are compared and discussed. Our future direction include the combination of these two methods for binding mode prediction.
Keywords: Drug discovery; Molecular docking; Molecular similarity; Protein–ligand interaction; Template-based.
Figures

References
-
- Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3:935–947. - PubMed
-
- Brooijmans N, Kuntz ID. (2003) Molecular recognition and docking algorithms. Annual review of biophysics and biomolecular structure. 2003 June;32(1):335–373. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
