

Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis
- PMID: 30692209
- PMCID: PMC6362813
- DOI: 10.1101/gad.320481.118
Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis
Abstract
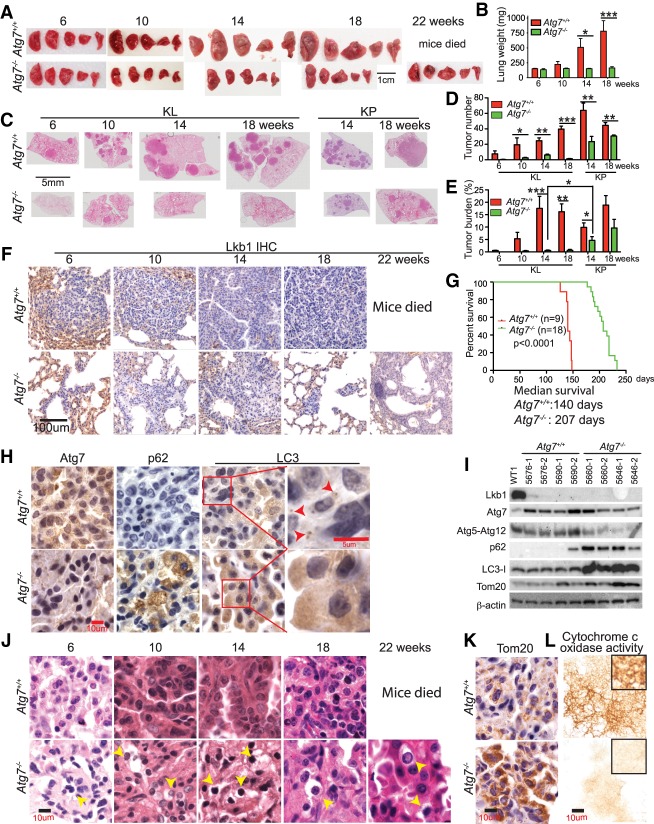
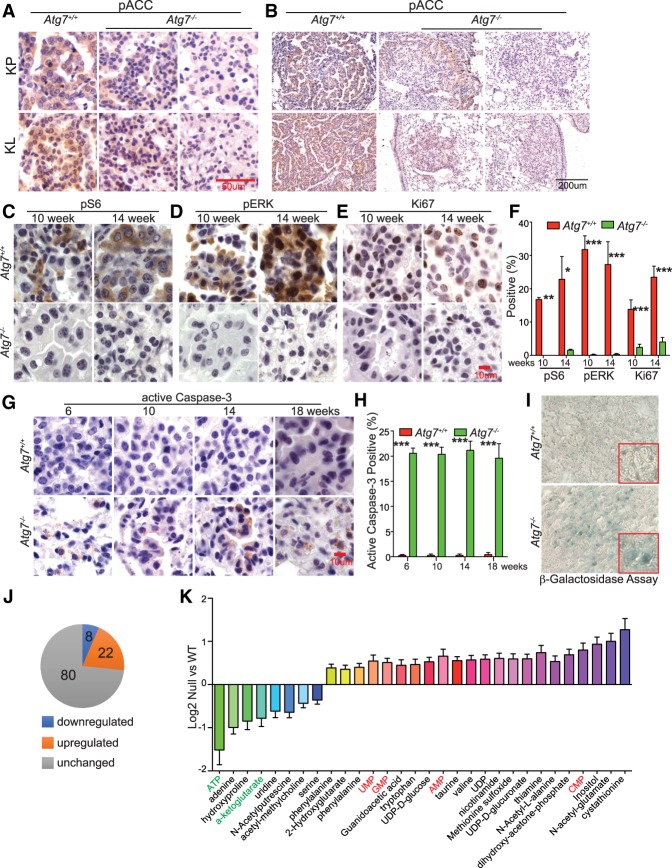
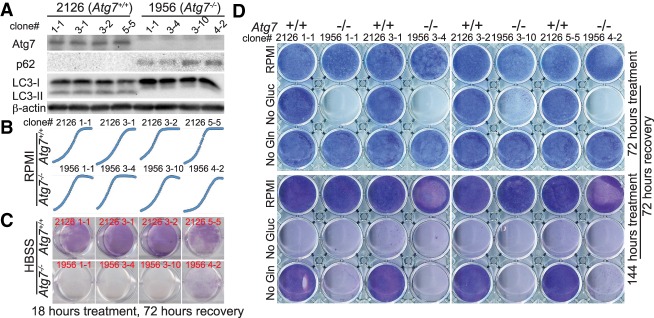
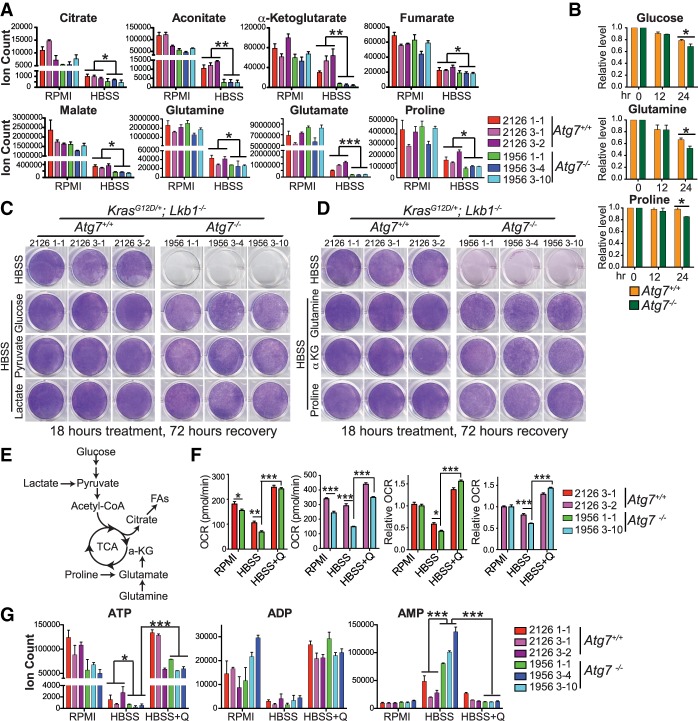
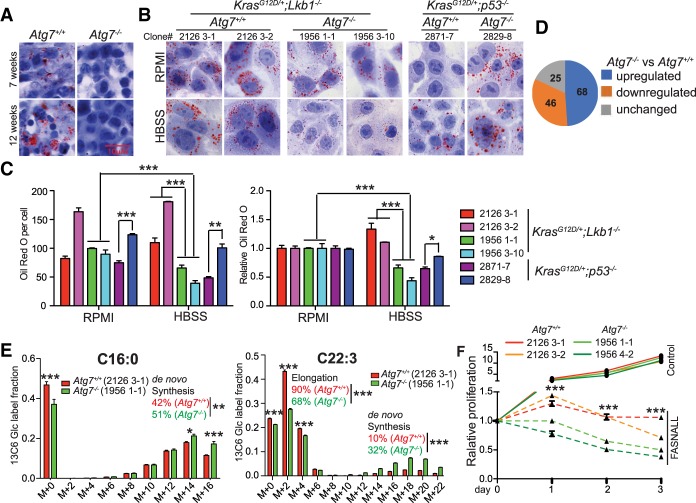
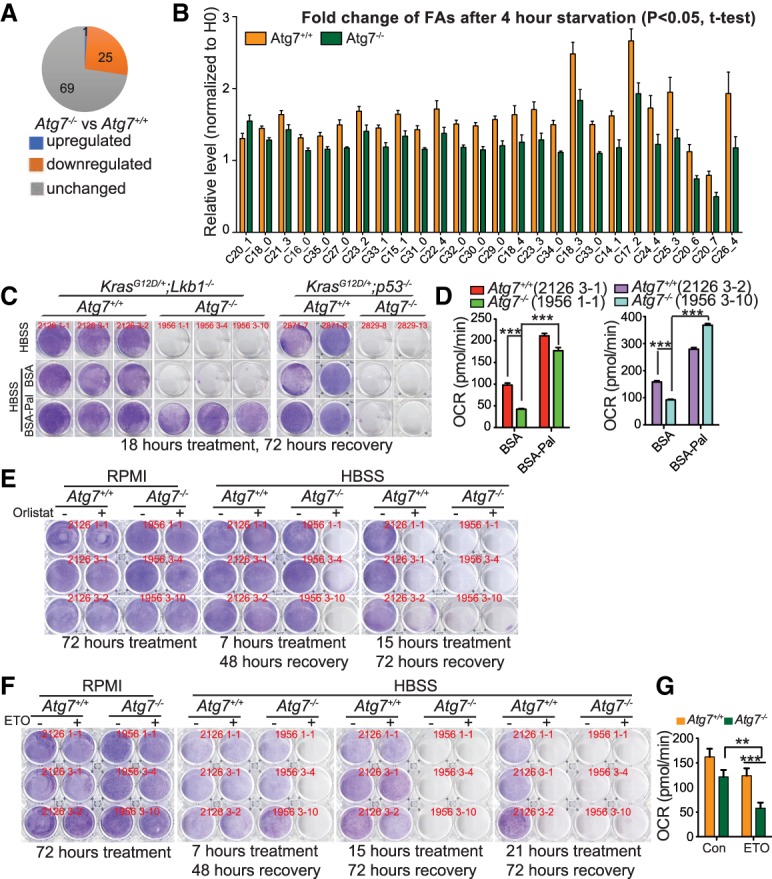
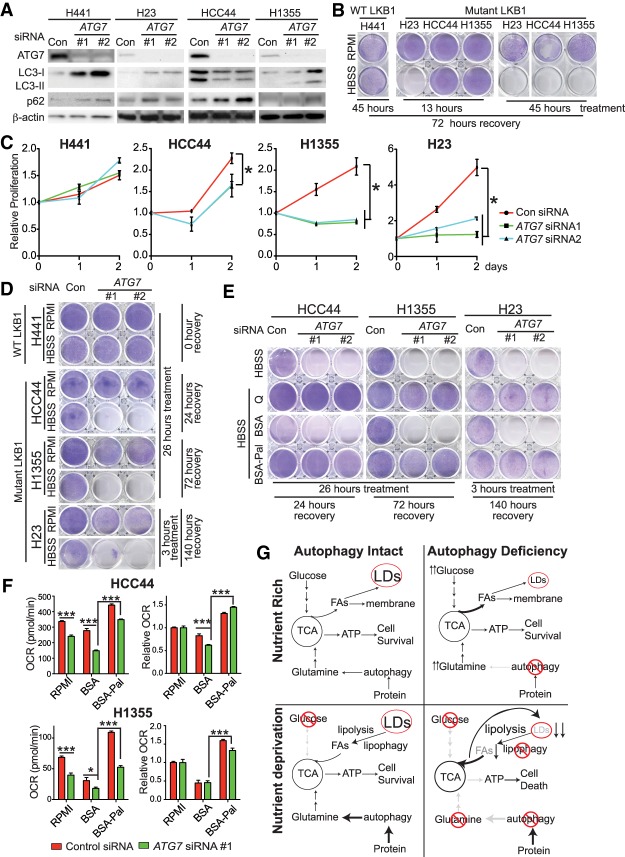
Loss of tumor suppressor liver kinase B1 (LKB1) promotes cancer cell proliferation but also leads to decreased metabolic plasticity in dealing with energy crises. Autophagy is a protective process involving self-cannibalization to maintain cellular energy homeostasis during nutrient deprivation. We developed a mouse model for Lkb1-deficient lung cancer with conditional deletion of essential autophagy gene Atg7 to test whether autophagy compensates for LKB1 loss for tumor cells to survive energy crises. We found that autophagy ablation was synthetically lethal during Lkb1-deficient lung tumorigenesis in both tumor initiation and tumor growth. We further found that autophagy deficiency causes defective intracellular recycling, which limits amino acids to support mitochondrial energy production in starved cancer cells and causes autophagy-deficient cells to be more dependent on fatty acid oxidation (FAO) for energy production, leading to reduced lipid reserve and energy crisis. Our findings strongly suggest that autophagy inhibition could be a strategy for treating LKB1-deficient lung tumors.
Keywords: LKB1; autophagy; energy metabolism; lipid metabolism; non-small cell lung cancer.
© 2019 Bhatt et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous