

Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity
- PMID: 30692544
- PMCID: PMC6349937
- DOI: 10.1038/s41467-018-08205-7
Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity
Abstract
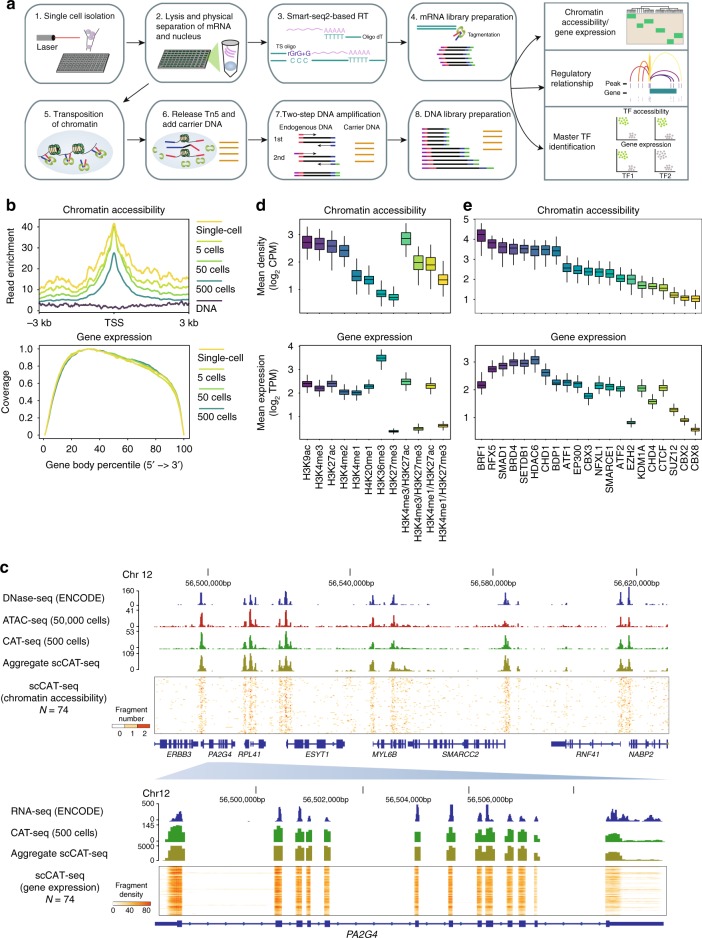
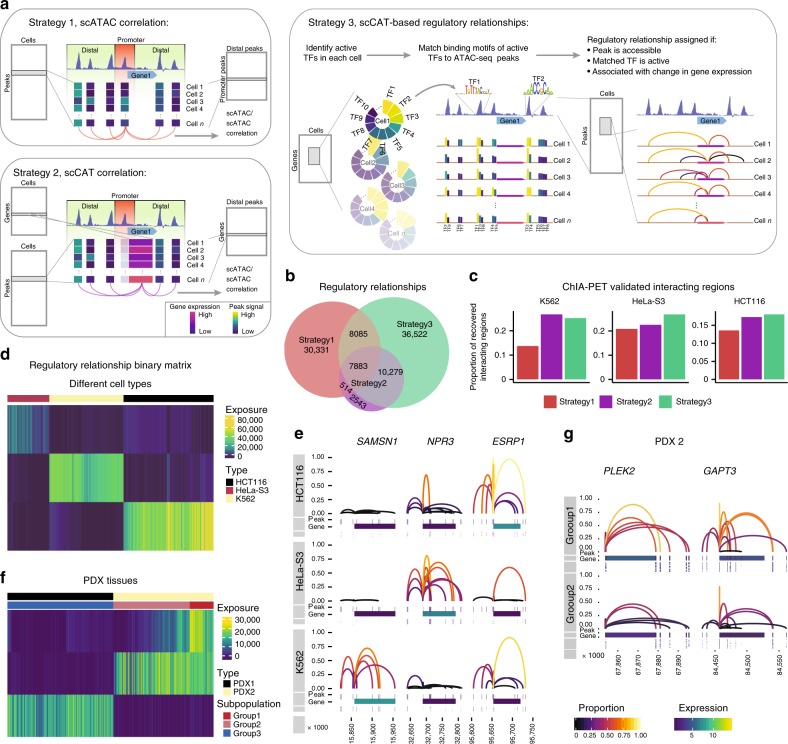
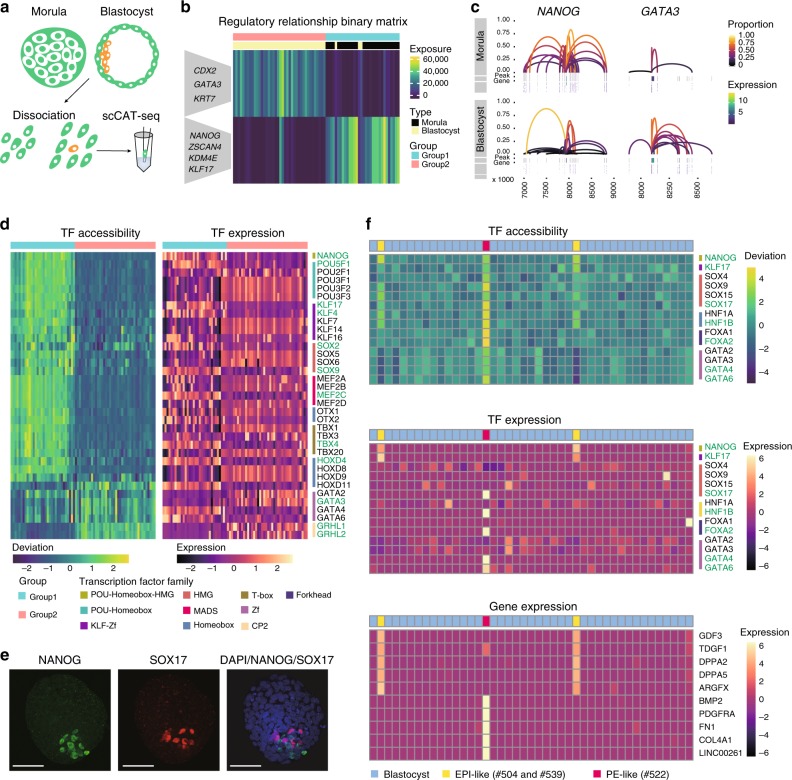
Integrative analysis of multi-omics layers at single cell level is critical for accurate dissection of cell-to-cell variation within certain cell populations. Here we report scCAT-seq, a technique for simultaneously assaying chromatin accessibility and the transcriptome within the same single cell. We show that the combined single cell signatures enable accurate construction of regulatory relationships between cis-regulatory elements and the target genes at single-cell resolution, providing a new dimension of features that helps direct discovery of regulatory patterns specific to distinct cell identities. Moreover, we generate the first single cell integrated map of chromatin accessibility and transcriptome in early embryos and demonstrate the robustness of scCAT-seq in the precise dissection of master transcription factors in cells of distinct states. The ability to obtain these two layers of omics data will help provide more accurate definitions of "single cell state" and enable the deconvolution of regulatory heterogeneity from complex cell populations.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
