

ApoE attenuates unresolvable inflammation by complex formation with activated C1q
- PMID: 30692699
- PMCID: PMC6420126
- DOI: 10.1038/s41591-018-0336-8
ApoE attenuates unresolvable inflammation by complex formation with activated C1q
Erratum in
-
Publisher Correction: ApoE attenuates unresolvable inflammation by complex formation with activated C1q.Nat Med. 2019 Mar;25(3):529. doi: 10.1038/s41591-019-0378-6. Nat Med. 2019. PMID: 30718908
Abstract
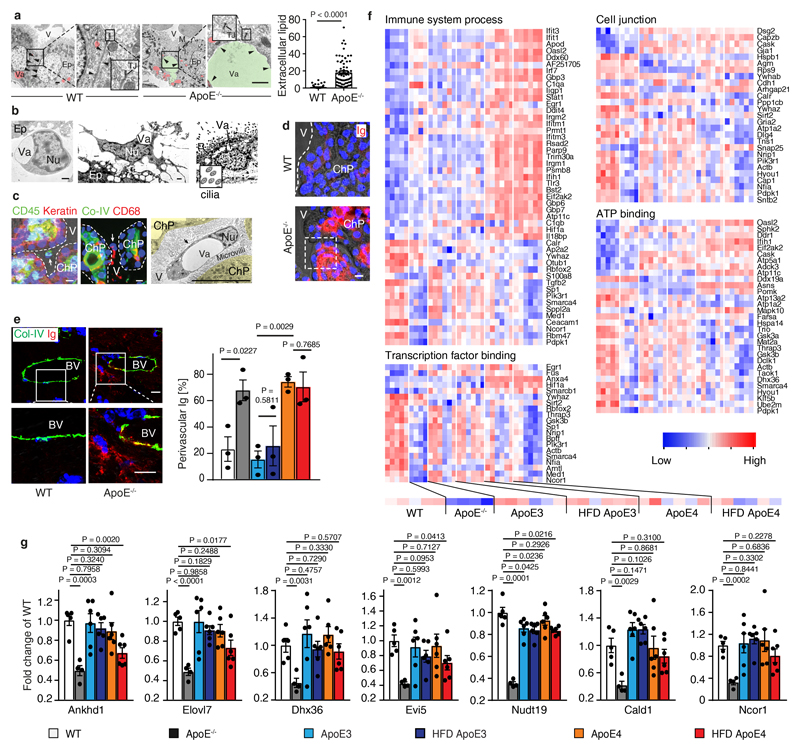
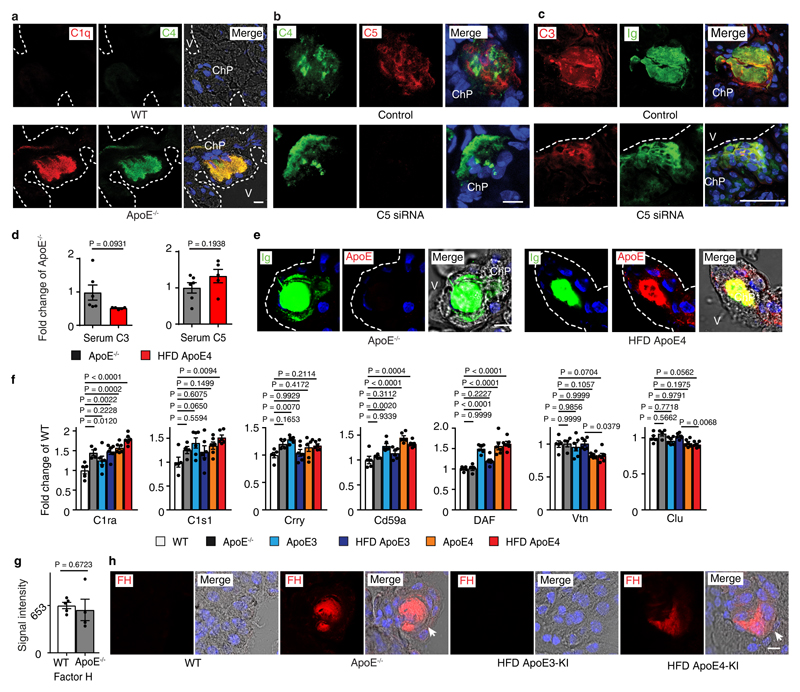
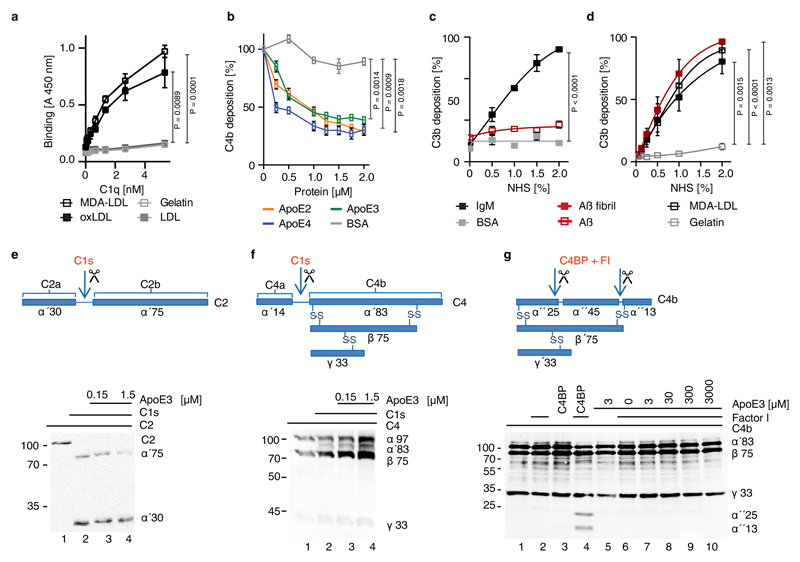
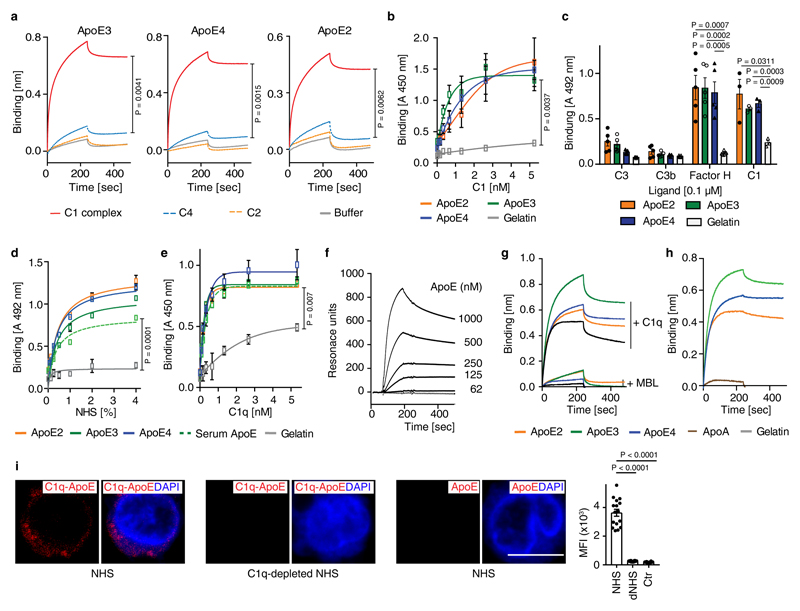
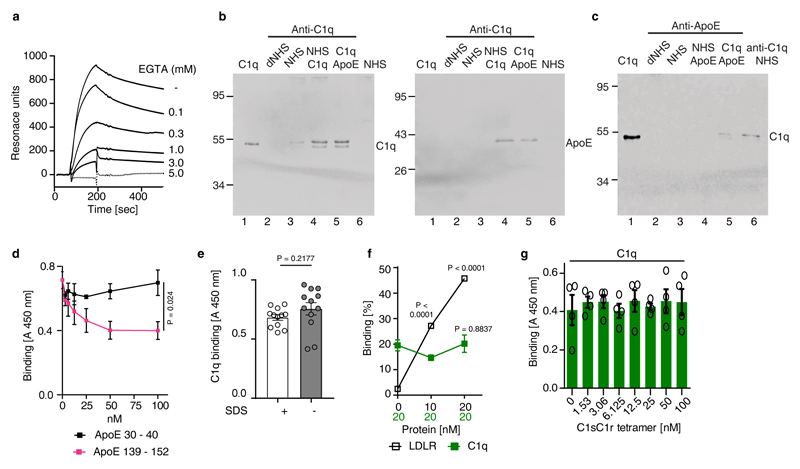
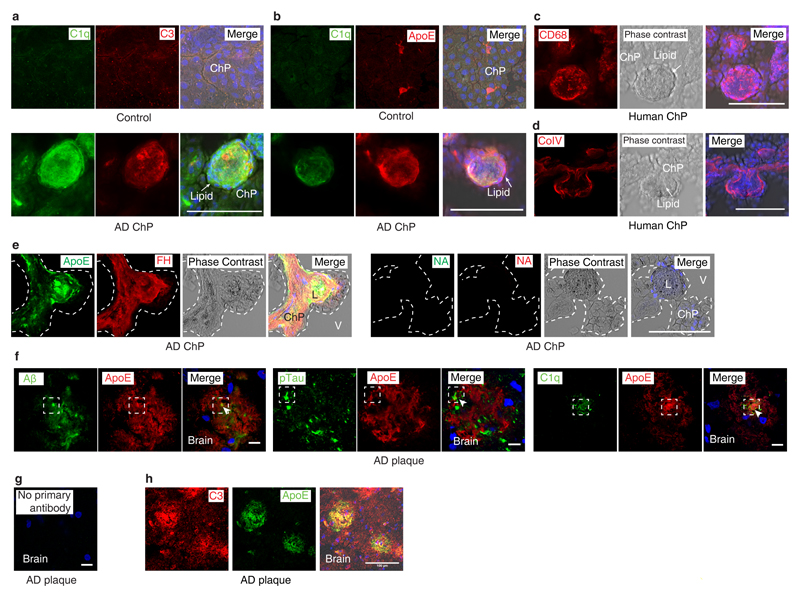
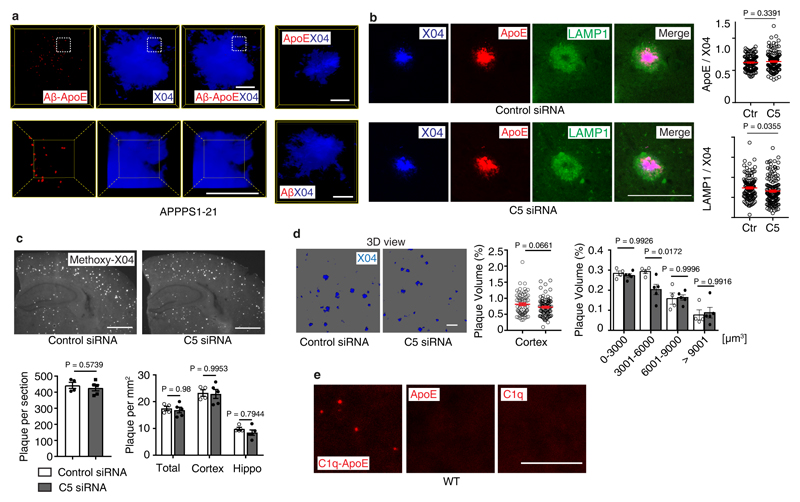
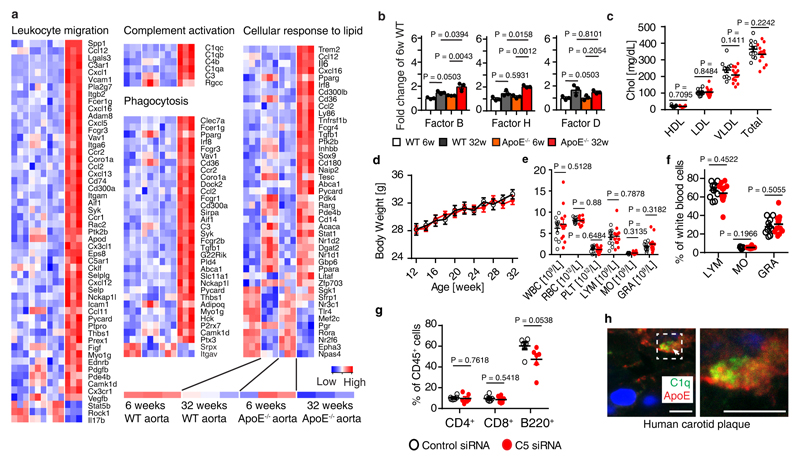
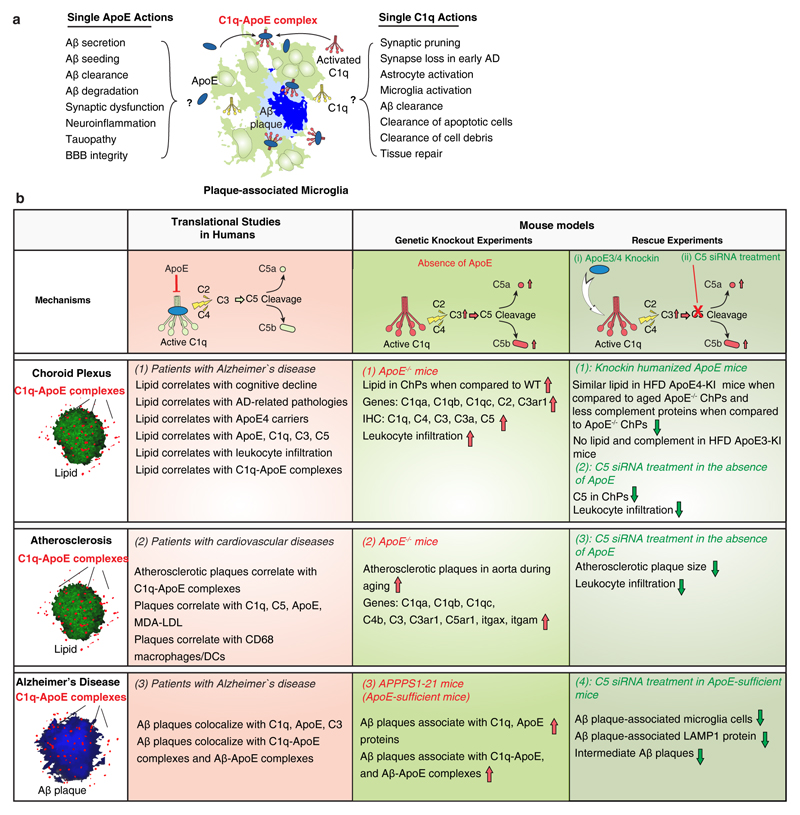
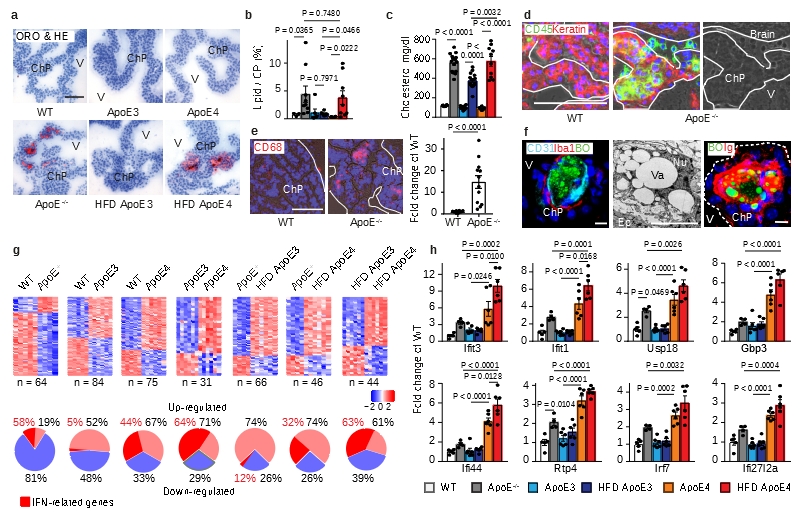
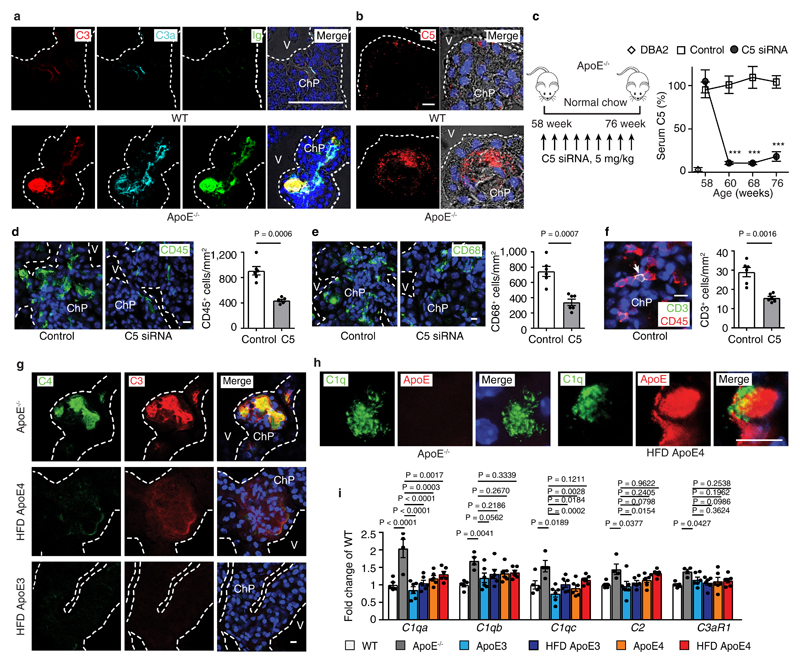
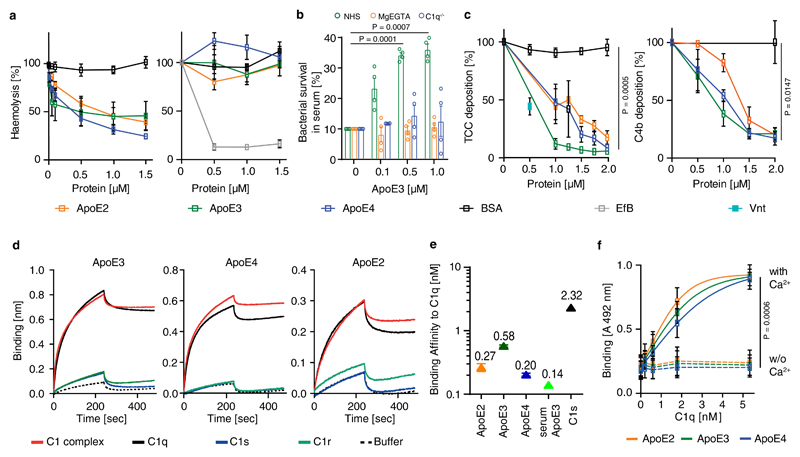
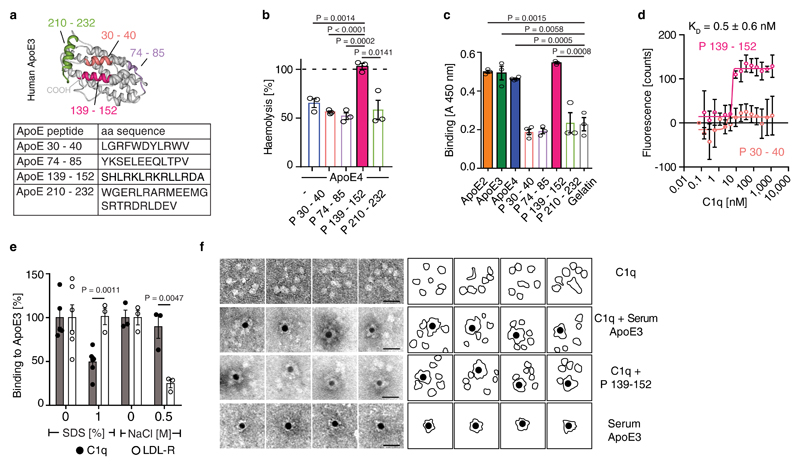
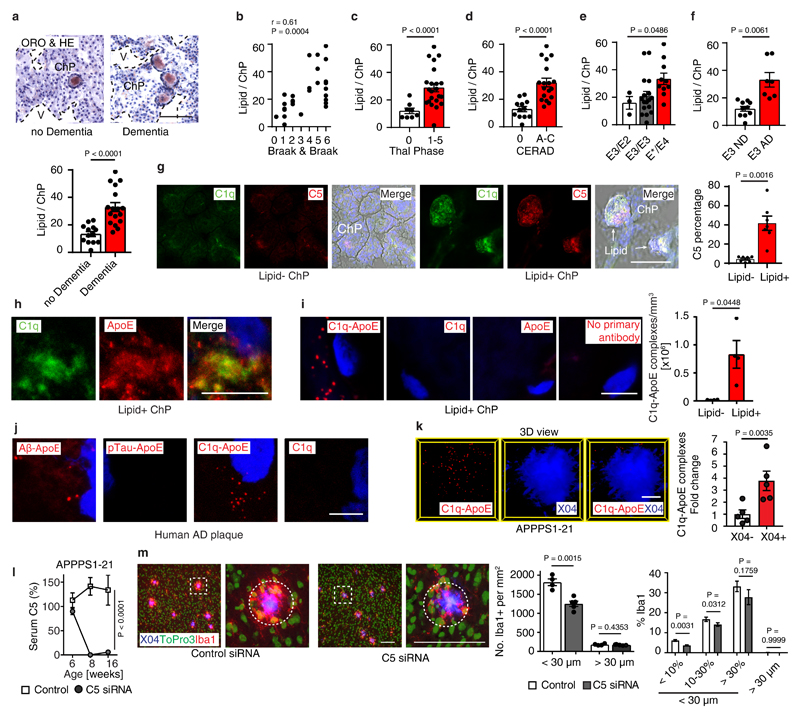
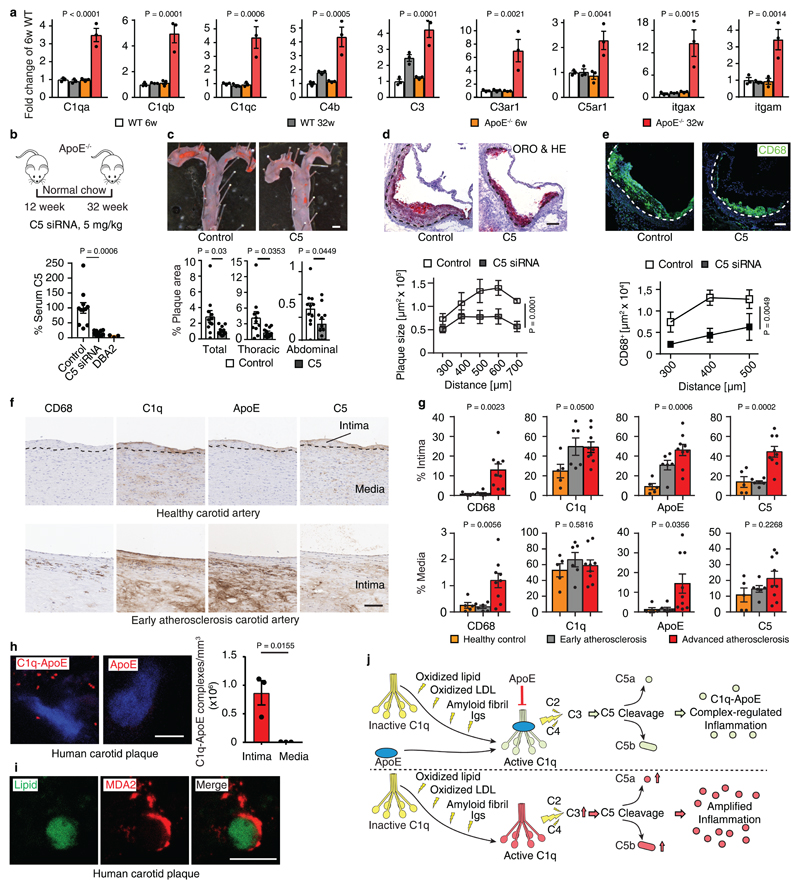
Apolipoprotein-E (ApoE) has been implicated in Alzheimer's disease, atherosclerosis, and other unresolvable inflammatory conditions but a common mechanism of action remains elusive. We found in ApoE-deficient mice that oxidized lipids activated the classical complement cascade (CCC), resulting in leukocyte infiltration of the choroid plexus (ChP). All human ApoE isoforms attenuated CCC activity via high-affinity binding to the activated CCC-initiating C1q protein (KD~140-580 pM) in vitro, and C1q-ApoE complexes emerged as markers for ongoing complement activity of diseased ChPs, Aβ plaques, and atherosclerosis in vivo. C1q-ApoE complexes in human ChPs, Aβ plaques, and arteries correlated with cognitive decline and atherosclerosis, respectively. Treatment with small interfering RNA (siRNA) against C5, which is formed by all complement pathways, attenuated murine ChP inflammation, Aβ-associated microglia accumulation, and atherosclerosis. Thus, ApoE is a direct checkpoint inhibitor of unresolvable inflammation, and reducing C5 attenuates disease burden.
Conflict of interest statement
C.Y., A.J.R.H., A.B., S.K.M., S.A., P.F.Z., and C.S. declare competing financial interests. C.Y., S.K.M., and A.J.R.H. are owners of Easemedcontrol R & D GmbH & Co KG Munich, Germany; A.B. is employed by Alnylam Pharmaceuticals Cambridge; Cambridge, MA, USA; C.Y. and A.J.R.H. have been named inventors on a pending patent application related to treatment and diagnosis of unresolvable inflammatory diseases (EP18183584.4); A.B. has been named as an inventor on patent applications related to C5 including PCT publication WO2014160129, and applications and patents based thereon; S.A., P.F.Z., and C.S. have been named inventors on a pending patent application (DE 10 2018 100 377.3).
Figures
Comment in
-
Resolving atherosclerosis and Alzheimer disease.Nat Rev Cardiol. 2019 May;16(5):259-260. doi: 10.1038/s41569-019-0182-5. Nat Rev Cardiol. 2019. PMID: 30903022 No abstract available.
References
-
- Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science. 1995;267:1034–1037. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
