

The BRUCE-ATR Signaling Axis Is Required for Accurate DNA Replication and Suppression of Liver Cancer Development
- PMID: 30693543
- PMCID: PMC6541504
- DOI: 10.1002/hep.30529
The BRUCE-ATR Signaling Axis Is Required for Accurate DNA Replication and Suppression of Liver Cancer Development
Abstract
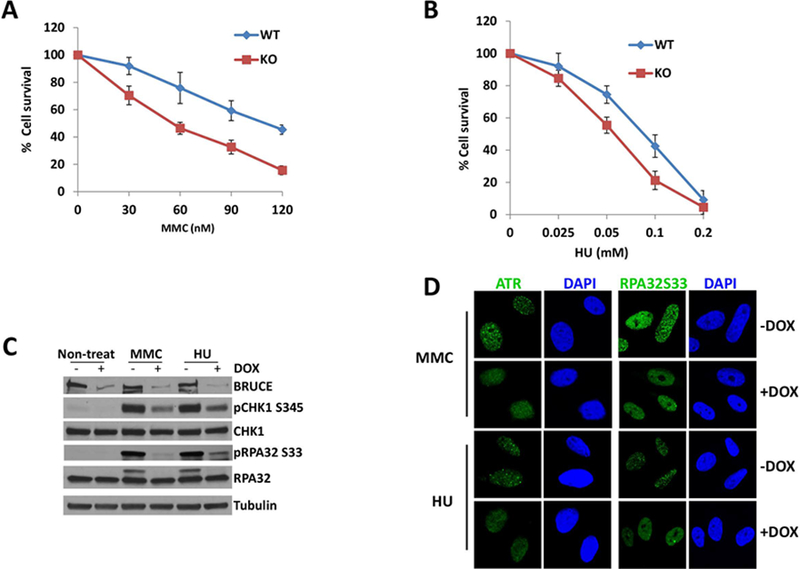
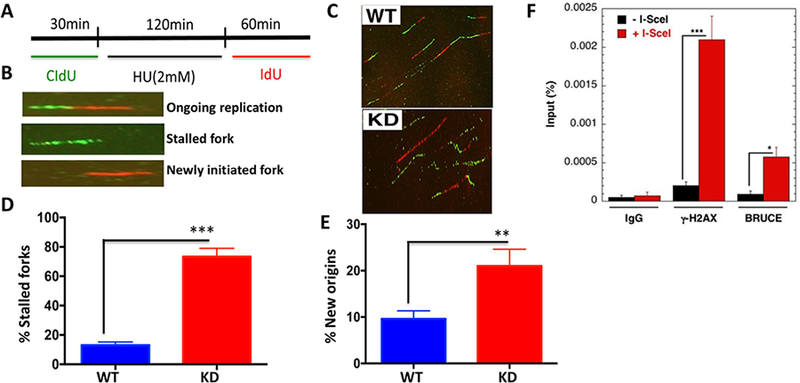
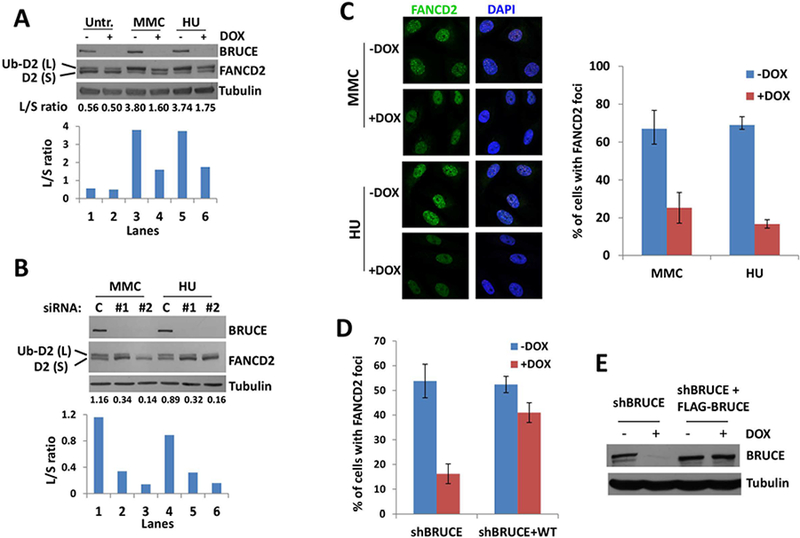
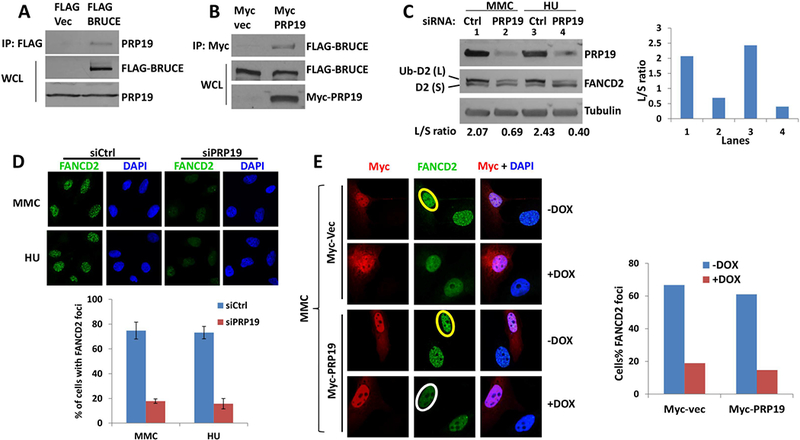
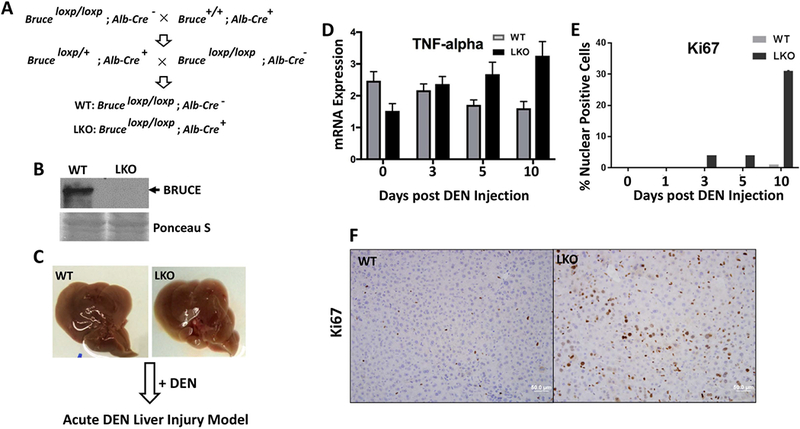
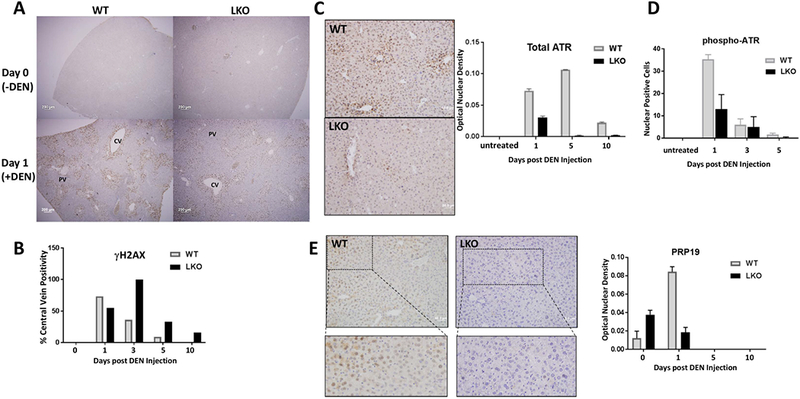
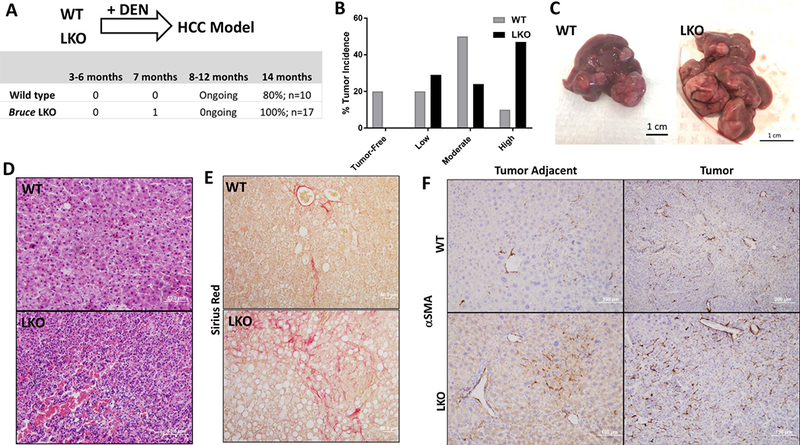
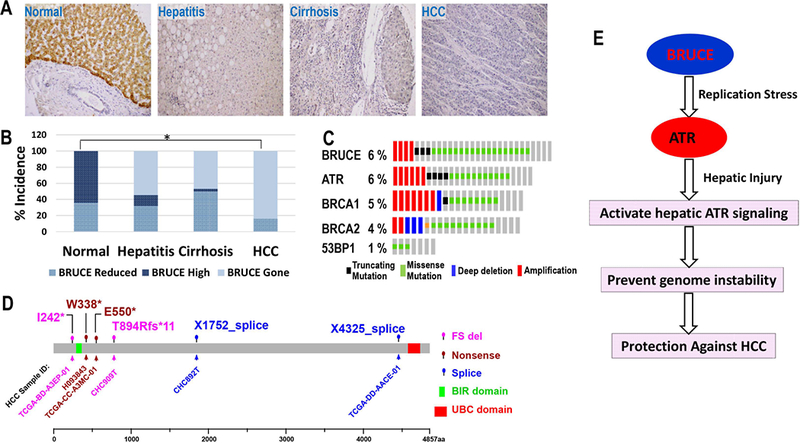
Replication fork stability during DNA replication is vital for maintenance of genomic stability and suppression of cancer development in mammals. ATR (ataxia-telangiectasia mutated [ATM] and RAD3-related) is a master regulatory kinase that activates the replication stress response to overcome replication barriers. Although many downstream effectors of ATR have been established, the upstream regulators of ATR and the effect of such regulation on liver cancer remain unclear. The ubiquitin conjugase BRUCE (BIR Repeat containing Ubiquitin-Conjugating Enzyme) is a guardian of chromosome integrity and activator of ATM signaling, which promotes DNA double-strand break repair through homologous recombination. Here we demonstrate the functions for BRUCE in ATR activation in vitro and liver tumor suppression in vivo. BRUCE is recruited to induced DNA damage sites. Depletion of BRUCE inhibited multiple ATR-dependent signaling events during replication stress, including activation of ATR itself, phosphorylation of its downstream targets CHK1 and RPA, and the mono-ubiquitination of FANCD2. Consequently, BRUCE deficiency resulted in stalled DNA replication forks and increased firing of new replication origins. The in vivo impact of BRUCE loss on liver tumorigenesis was determined using the hepatocellular carcinoma model induced by genotoxin diethylnitrosamine. Liver-specific knockout of murine Bruce impaired ATR activation and exacerbated inflammation, fibrosis and hepatocellular carcinoma, which exhibited a trabecular architecture, closely resembling human hepatocellular carcinoma (HCC). In humans, the clinical relevance of BRUCE down-regulation in liver disease was found in hepatitis, cirrhosis, and HCC specimens, and deleterious somatic mutations of the Bruce gene was found in human hepatocellular carcinoma in the Cancer Genome Atlas database. Conclusion: These findings establish a BRUCE-ATR signaling axis in accurate DNA replication and suppression of liver cancer in mice and humans and provides a clinically relevant HCC mouse model.
© 2019 by the American Association for the Study of Liver Diseases.
Conflict of interest statement
Figures
References
-
- Chen Z, Naito M, Hori S, Mashima T, Yamori T, Tsuruo T. A human IAP-family gene, apollon, expressed in human brain cancer cells. Biochem Biophys Res Commun 1999;264:847–854. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
