Machine Learning and Integrative Analysis of Biomedical Big Data
- PMID: 30696086
- PMCID: PMC6410075
- DOI: 10.3390/genes10020087
Machine Learning and Integrative Analysis of Biomedical Big Data
Abstract
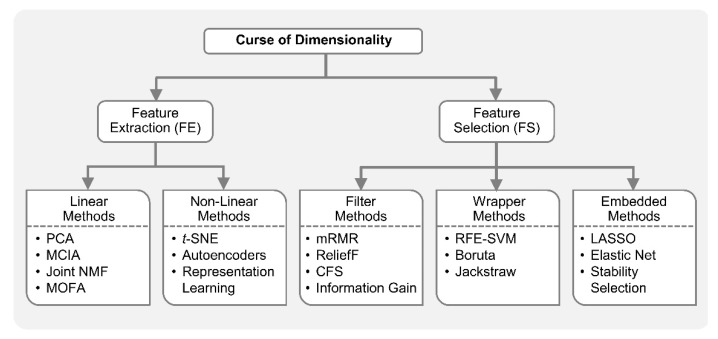
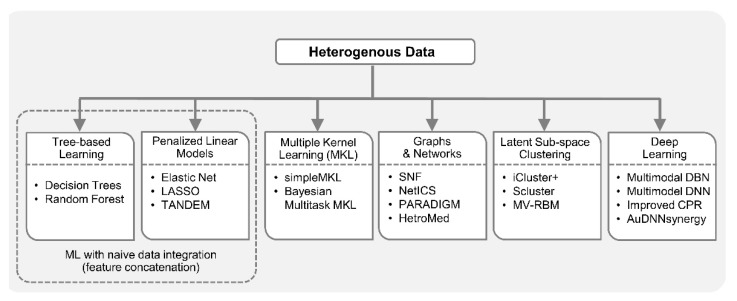
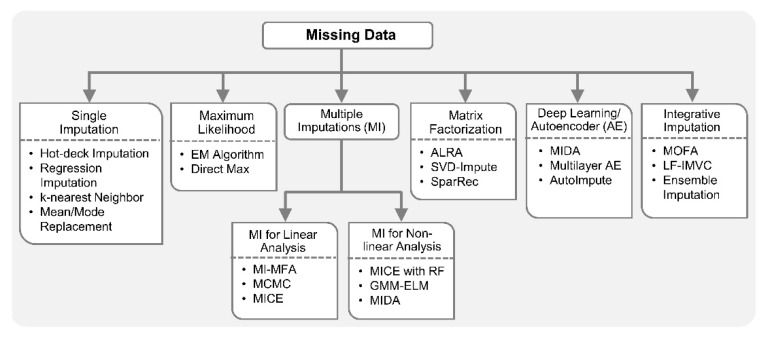
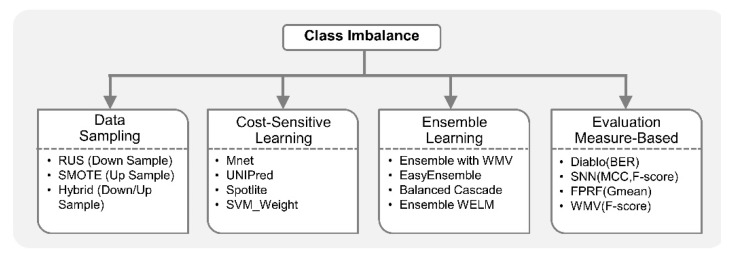
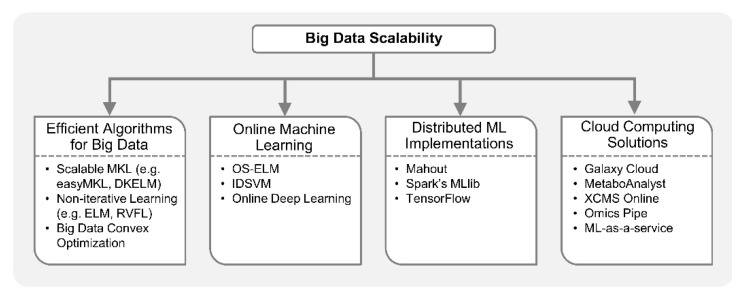
Recent developments in high-throughput technologies have accelerated the accumulation of massive amounts of omics data from multiple sources: genome, epigenome, transcriptome, proteome, metabolome, etc. Traditionally, data from each source (e.g., genome) is analyzed in isolation using statistical and machine learning (ML) methods. Integrative analysis of multi-omics and clinical data is key to new biomedical discoveries and advancements in precision medicine. However, data integration poses new computational challenges as well as exacerbates the ones associated with single-omics studies. Specialized computational approaches are required to effectively and efficiently perform integrative analysis of biomedical data acquired from diverse modalities. In this review, we discuss state-of-the-art ML-based approaches for tackling five specific computational challenges associated with integrative analysis: curse of dimensionality, data heterogeneity, missing data, class imbalance and scalability issues.
Keywords: class imbalance; curse of dimensionality; data integration; heterogeneous data; machine learning; missing data; multi-omics; scalability.
Conflict of interest statement
The authors declare no conflict of interest.
Figures