

Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy
- PMID: 30696458
- PMCID: PMC6350371
- DOI: 10.1186/s13073-019-0616-z
Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy
Abstract
Background: International guidelines for variant interpretation in Mendelian disease set stringent criteria to report a variant as (likely) pathogenic, prioritising control of false-positive rate over test sensitivity and diagnostic yield. Genetic testing is also more likely informative in individuals with well-characterised variants from extensively studied European-ancestry populations. Inherited cardiomyopathies are relatively common Mendelian diseases that allow empirical calibration and assessment of this framework.
Methods: We compared rare variants in large hypertrophic cardiomyopathy (HCM) cohorts (up to 6179 cases) to reference populations to identify variant classes with high prior likelihoods of pathogenicity, as defined by etiological fraction (EF). We analysed the distribution of variants using a bespoke unsupervised clustering algorithm to identify gene regions in which variants are significantly clustered in cases.
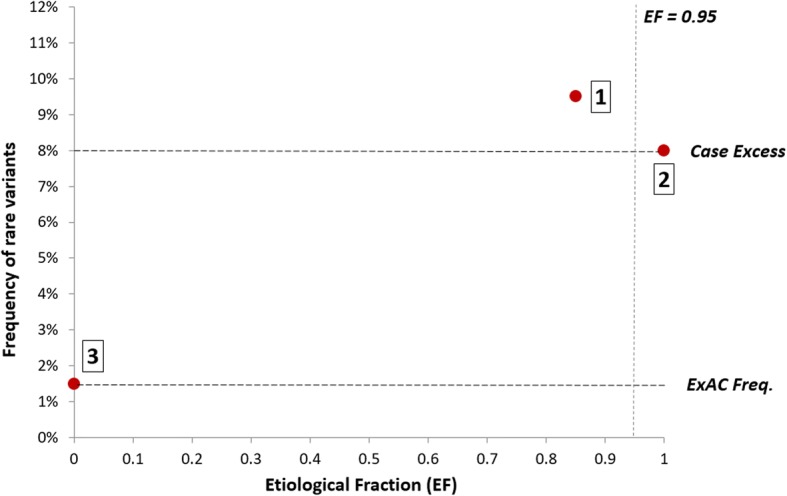
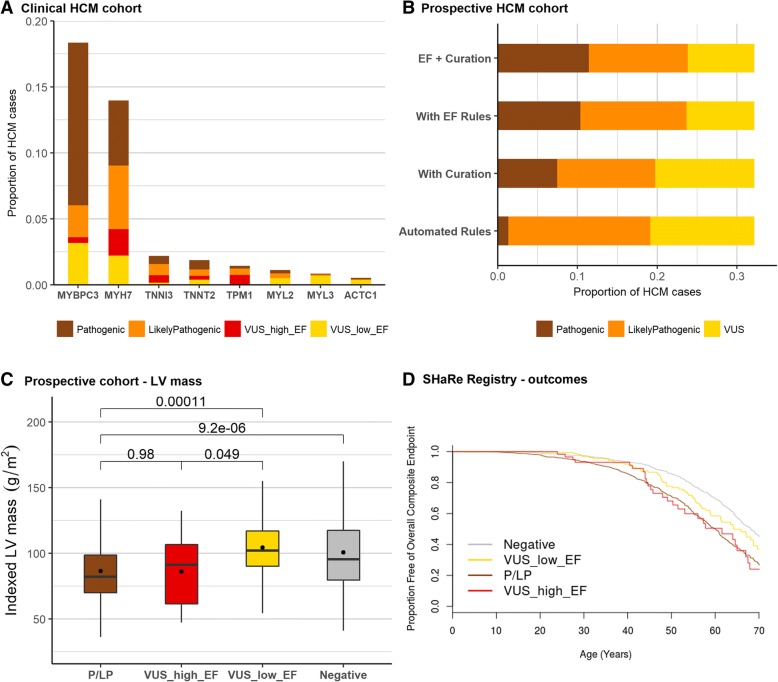
Results: Analysis of variant distribution identified regions in which variants are significantly enriched in cases and variant location was a better discriminator of pathogenicity than generic computational functional prediction algorithms. Non-truncating variant classes with an EF ≥ 0.95 were identified in five established HCM genes. Applying this approach leads to an estimated 14-20% increase in cases with actionable HCM variants, i.e. variants classified as pathogenic/likely pathogenic that might be used for predictive testing in probands' relatives.
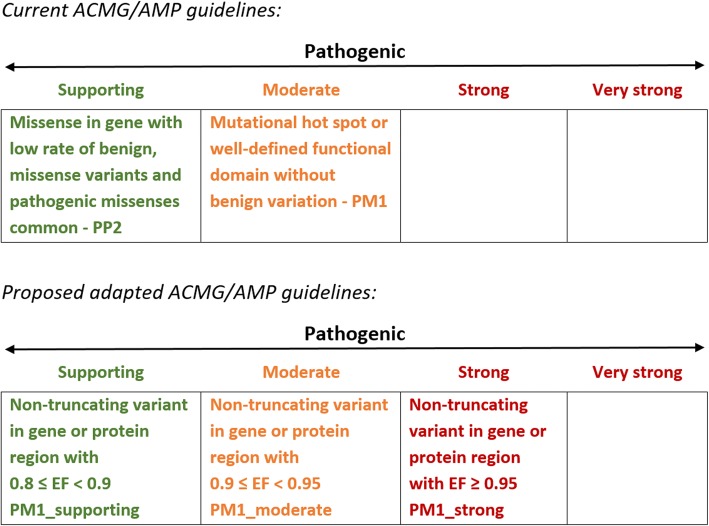
Conclusions: When found in a patient confirmed to have disease, novel variants in some genes and regions are empirically shown to have a sufficiently high probability of pathogenicity to support a "likely pathogenic" classification, even without additional segregation or functional data. This could increase the yield of high confidence actionable variants, consistent with the framework and recommendations of current guidelines. The techniques outlined offer a consistent and unbiased approach to variant interpretation for Mendelian disease genetic testing. We propose adaptations to ACMG/AMP guidelines to incorporate such evidence in a quantitative and transparent manner.
Keywords: ACMG/AMP guidelines; Hypertrophic cardiomyopathy; Mendelian genetics; Variant interpretation.
Conflict of interest statement
Ethics approval and consent to participate
This study was based on previously published clinical genetics sequencing data from the OMGL and LMM laboratories. For the prospective HCM cohort from the Royal Brompton Hospital London, all patients provided written informed consent and the study was approved by the regional ethics committee. The research in this study conformed to the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


References
-
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. - DOI - PMC - PubMed
-
- Association for Clinical Genetic Science. Consensus statement on adoption of the American College of Medical Genetics and Genomics (ACMG) guidelines for sequence variant classification and interpretation [press release]. http://www.acgs.uk.com/media/1032817/acgs_consensus_statement_on_adoptio.... 2016.
