

SMCHD1 is involved in de novo methylation of the DUX4-encoding D4Z4 macrosatellite
- PMID: 30698748
- PMCID: PMC6451109
- DOI: 10.1093/nar/gkz005
SMCHD1 is involved in de novo methylation of the DUX4-encoding D4Z4 macrosatellite
Abstract
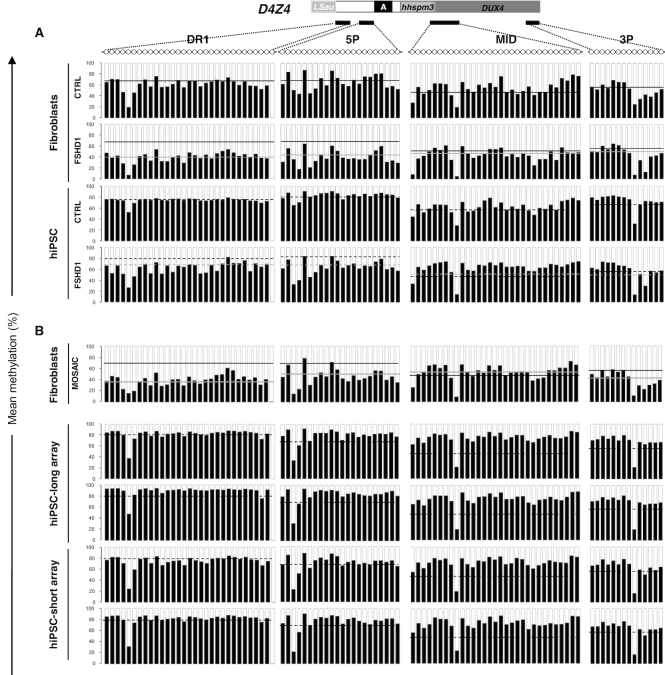
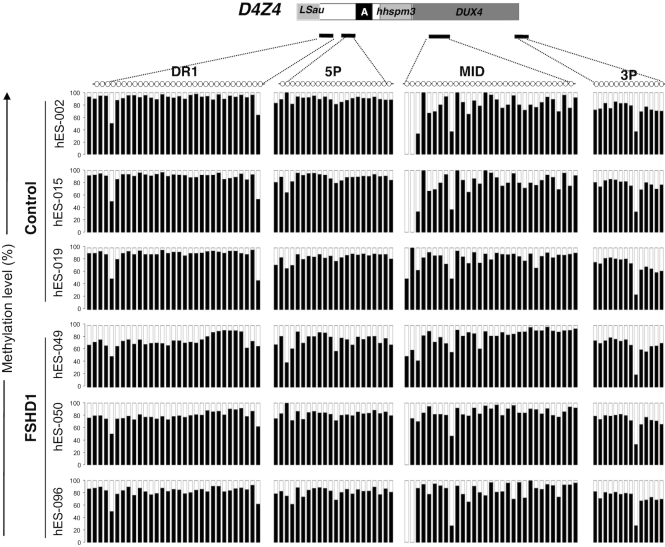
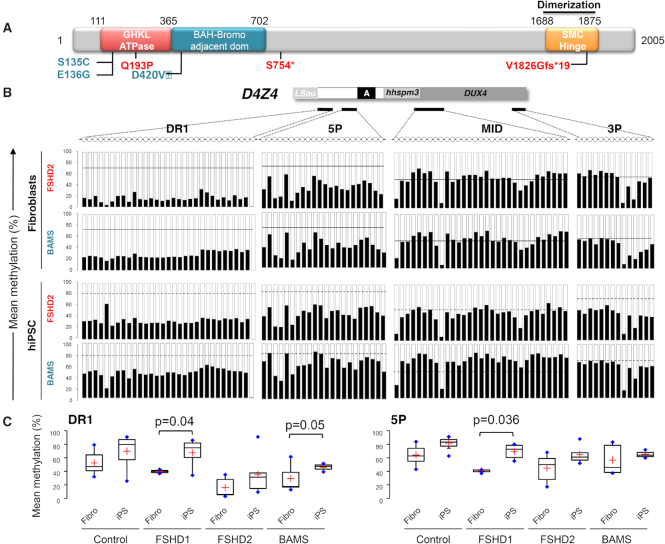
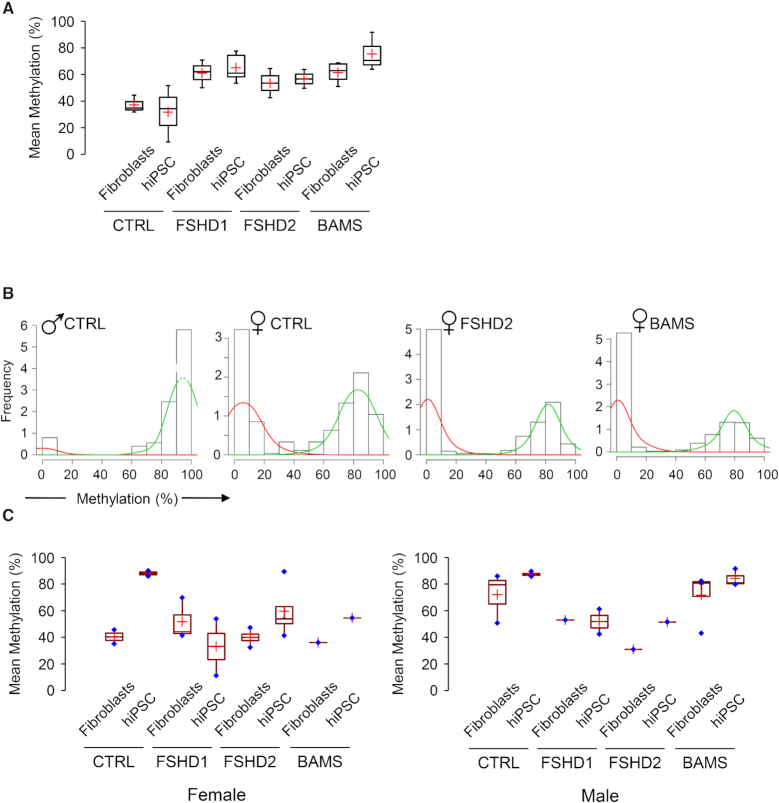
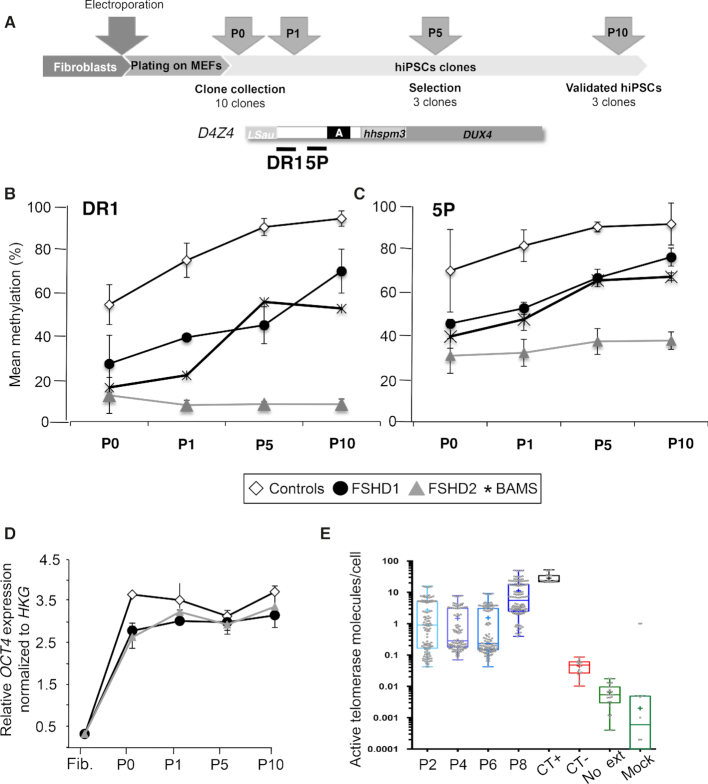
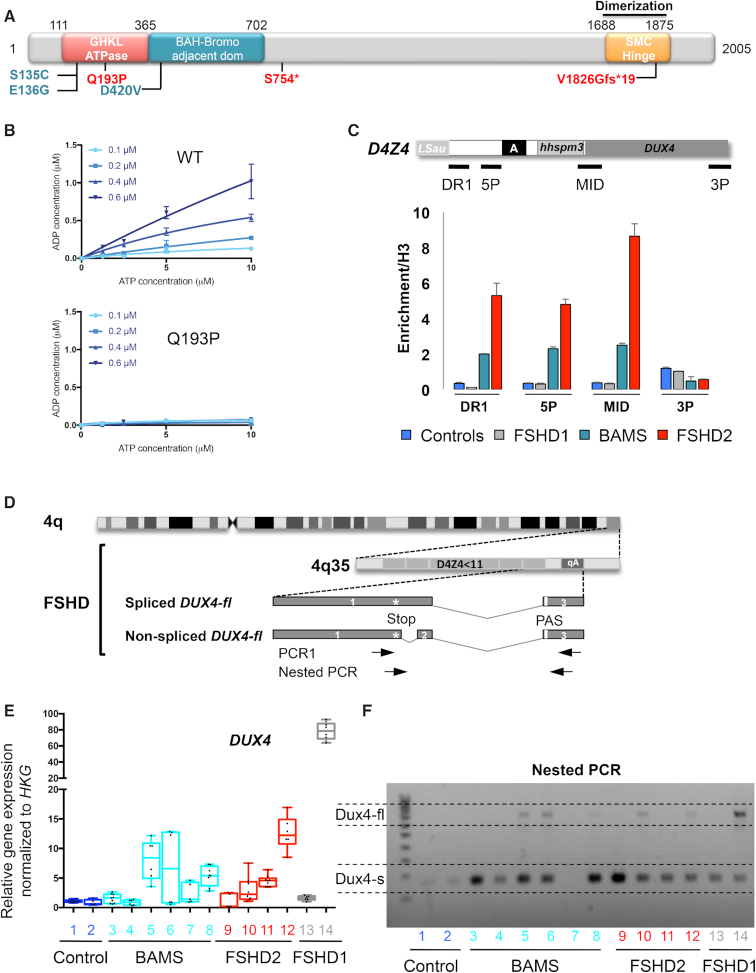
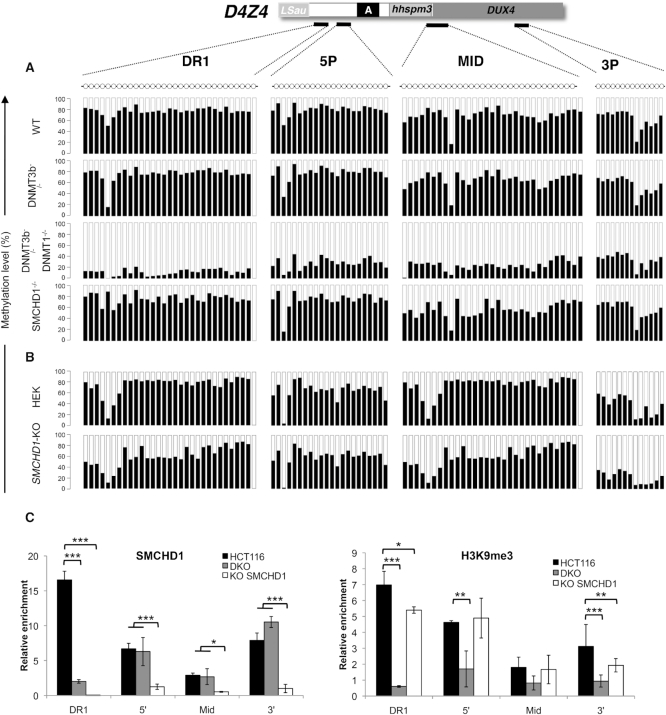
The DNA methylation epigenetic signature is a key determinant during development. Rules governing its establishment and maintenance remain elusive especially at repetitive sequences, which account for the majority of methylated CGs. DNA methylation is altered in a number of diseases including those linked to mutations in factors that modify chromatin. Among them, SMCHD1 (Structural Maintenance of Chromosomes Hinge Domain Containing 1) has been of major interest following identification of germline mutations in Facio-Scapulo-Humeral Dystrophy (FSHD) and in an unrelated developmental disorder, Bosma Arhinia Microphthalmia Syndrome (BAMS). By investigating why germline SMCHD1 mutations lead to these two different diseases, we uncovered a role for this factor in de novo methylation at the pluripotent stage. SMCHD1 is required for the dynamic methylation of the D4Z4 macrosatellite upon reprogramming but seems dispensable for methylation maintenance. We find that FSHD and BAMS patient's cells carrying SMCHD1 mutations are both permissive for DUX4 expression, a transcription factor whose regulation has been proposed as the main trigger for FSHD. These findings open new questions as to what is the true aetiology for FSHD, the epigenetic events associated with the disease thus calling the current model into question and opening new perspectives for understanding repetitive DNA sequences regulation.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Blewitt M.E., Gendrel A.V., Pang Z., Sparrow D.B., Whitelaw N., Craig J.M., Apedaile A., Hilton D.J., Dunwoodie S.L., Brockdorff N. et al.. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 2008; 40:663–669. - PubMed
-
- Chen K., Czabotar P.E., Blewitt M.E., Murphy J.M.. The hinge domain of the epigenetic repressor Smchd1 adopts an unconventional homodimeric configuration. Biochem. J. 2016; 473:733–742. - PubMed
-
- Jansz N., Chen K., Murphy J.M., Blewitt M.E.. The Epigenetic regulator SMCHD1 in development and disease. Trends Genet. 2017; 33:233–243. - PubMed
-
- Gendrel A.V., Apedaile A., Coker H., Termanis A., Zvetkova I., Godwin J., Tang Y.A., Huntley D., Montana G., Taylor S. et al.. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell. 2012; 23:265–279. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
