

Usability of reference-free transcriptome assemblies for detection of differential expression: a case study on Aethionema arabicum dimorphic seeds
- PMID: 30700268
- PMCID: PMC6354389
- DOI: 10.1186/s12864-019-5452-4
Usability of reference-free transcriptome assemblies for detection of differential expression: a case study on Aethionema arabicum dimorphic seeds
Abstract
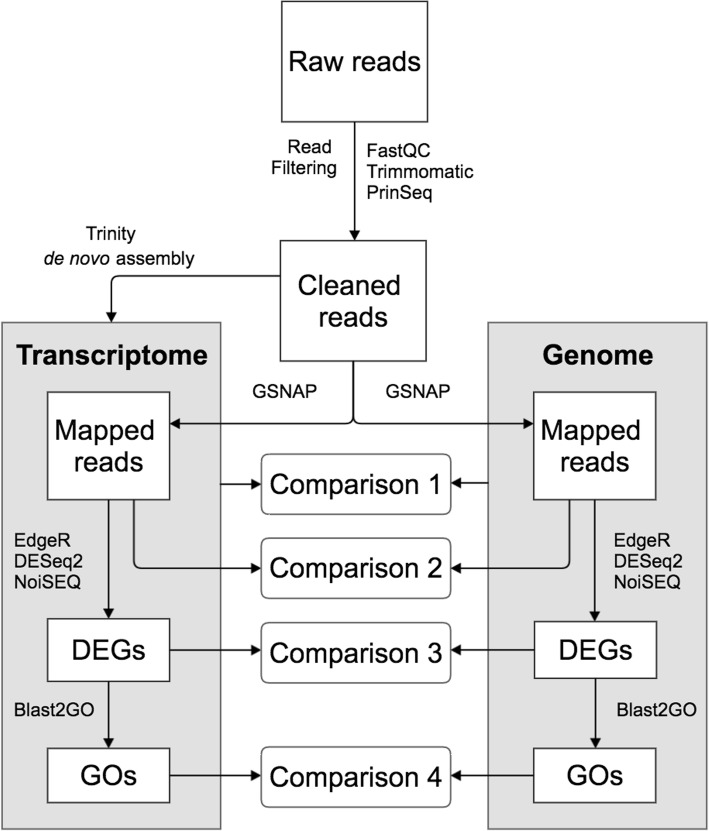
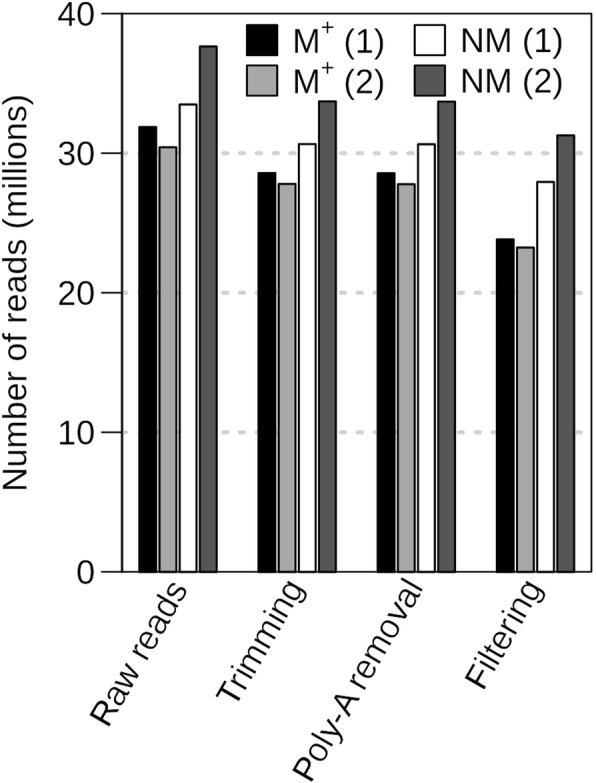
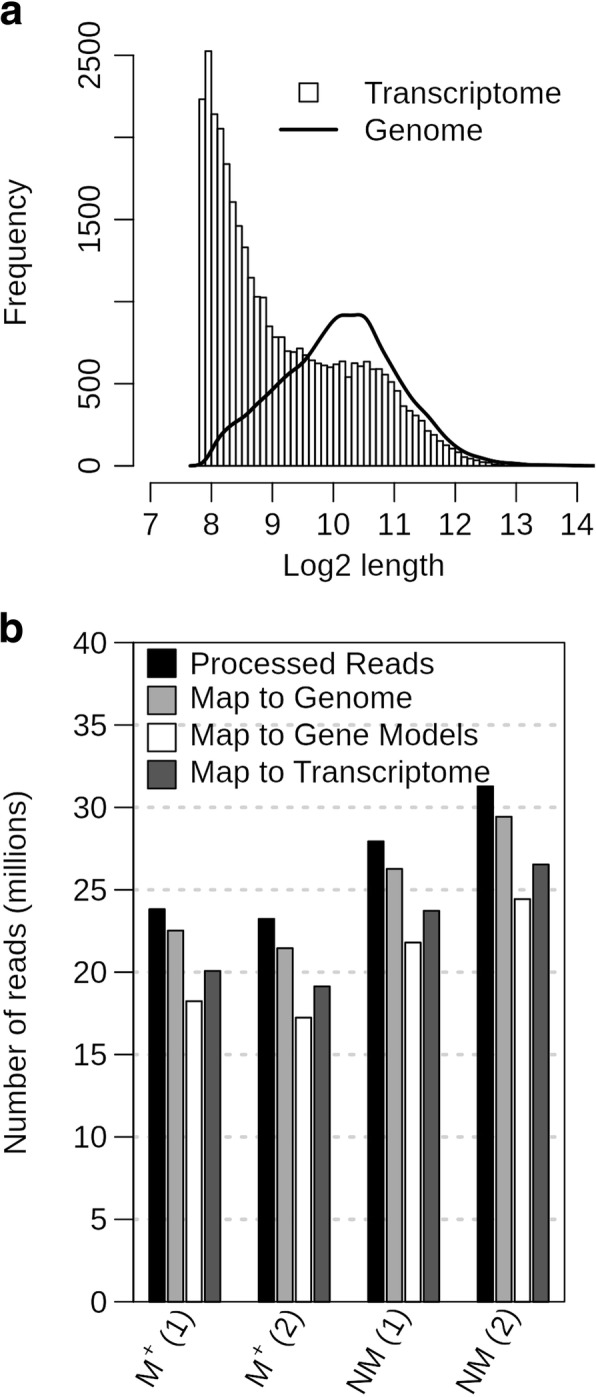
Background: RNA-sequencing analysis is increasingly utilized to study gene expression in non-model organisms without sequenced genomes. Aethionema arabicum (Brassicaceae) exhibits seed dimorphism as a bet-hedging strategy - producing both a less dormant mucilaginous (M+) seed morph and a more dormant non-mucilaginous (NM) seed morph. Here, we compared de novo and reference-genome based transcriptome assemblies to investigate Ae. arabicum seed dimorphism and to evaluate the reference-free versus -dependent approach for identifying differentially expressed genes (DEGs).
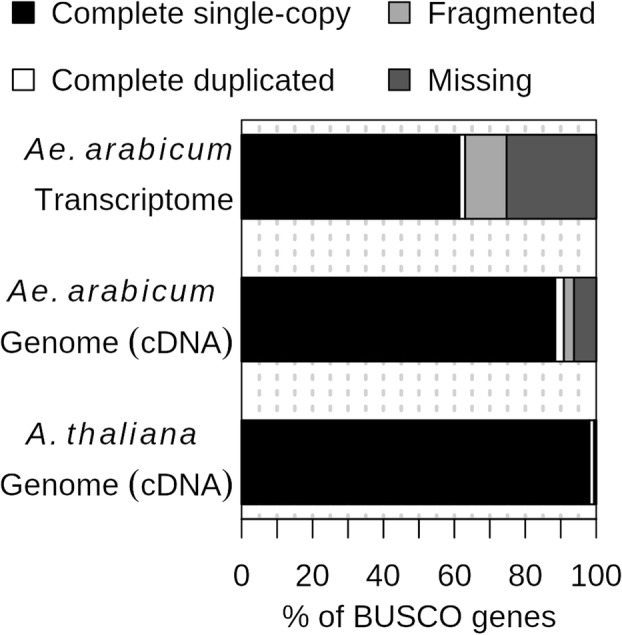
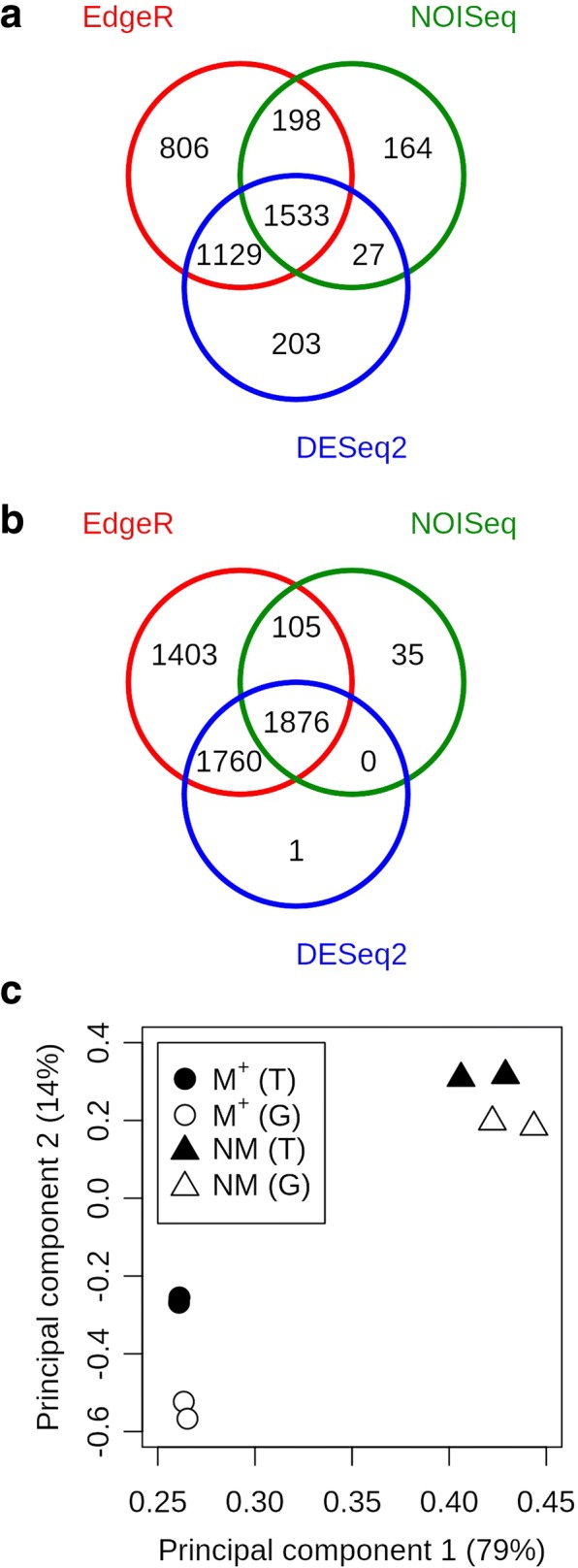
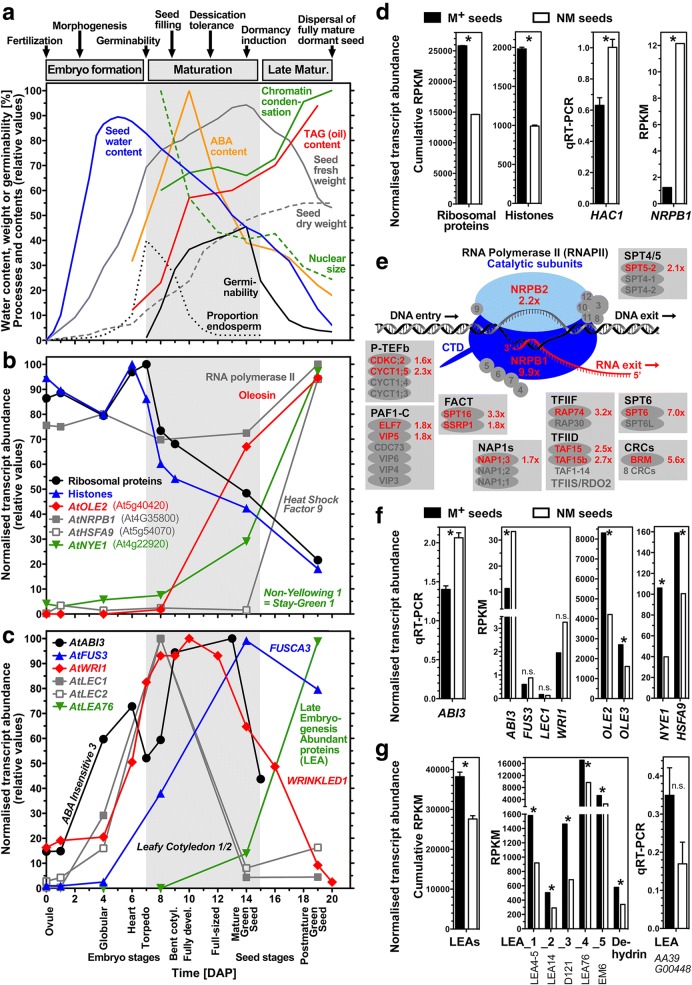
Results: A de novo transcriptome assembly was generated using sequences from M+ and NM Ae. arabicum dry seed morphs. The transcripts of the de novo assembly contained 63.1% complete Benchmarking Universal Single-Copy Orthologs (BUSCO) compared to 90.9% for the transcripts of the reference genome. DEG detection used the strict consensus of three methods (DESeq2, edgeR and NOISeq). Only 37% of 1533 differentially expressed de novo assembled transcripts paired with 1876 genome-derived DEGs. Gene Ontology (GO) terms distinguished the seed morphs: the terms translation and nucleosome assembly were overrepresented in DEGs higher in abundance in M+ dry seeds, whereas terms related to mRNA processing and transcription were overrepresented in DEGs higher in abundance in NM dry seeds. DEGs amongst these GO terms included ribosomal proteins and histones (higher in M+), RNA polymerase II subunits and related transcription and elongation factors (higher in NM). Expression of the inferred DEGs and other genes associated with seed maturation (e.g. those encoding late embryogenesis abundant proteins and transcription factors regulating seed development and maturation such as ABI3, FUS3, LEC1 and WRI1 homologs) were put in context with Arabidopsis thaliana seed maturation and indicated that M+ seeds may desiccate and mature faster than NM. The 1901 transcriptomic DEG set GO-terms had almost 90% overlap with the 2191 genome-derived DEG GO-terms.
Conclusions: Whilst there was only modest overlap of DEGs identified in reference-free versus -dependent approaches, the resulting GO analysis was concordant in both approaches. The identified differences in dry seed transcriptomes suggest mechanisms underpinning previously identified contrasts between morphology and germination behaviour of M+ and NM seeds.
Keywords: Aethionema arabicum; Dimorphic seeds; RNA-seq; Reference and reference-free; Transcriptome.
Conflict of interest statement
Ethics approval and consent to participate
The source of the
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures


References
-
- Lenser T, Graeber K, Cevik OS, Adiguzel N, Donmez AA, Grosche C, Kettermann M, Mayland-Quellhorst S, Merai Z, Mohammadin S, et al. Developmental control and plasticity of fruit and seed dimorphism in Aethionema arabicum. Plant Physiol. 2016;172(3):1691–1707. doi: 10.1104/pp.16.00838. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
