

A homozygous missense variant in the alkaline phosphatase gene ALPL is associated with a severe form of canine hypophosphatasia
- PMID: 30700765
- PMCID: PMC6353930
- DOI: 10.1038/s41598-018-37801-2
A homozygous missense variant in the alkaline phosphatase gene ALPL is associated with a severe form of canine hypophosphatasia
Abstract
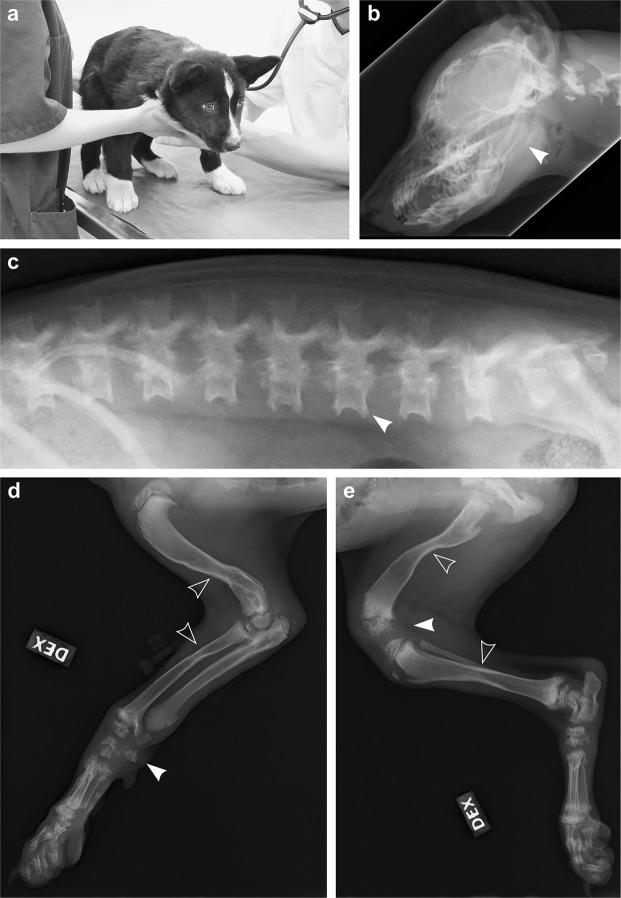
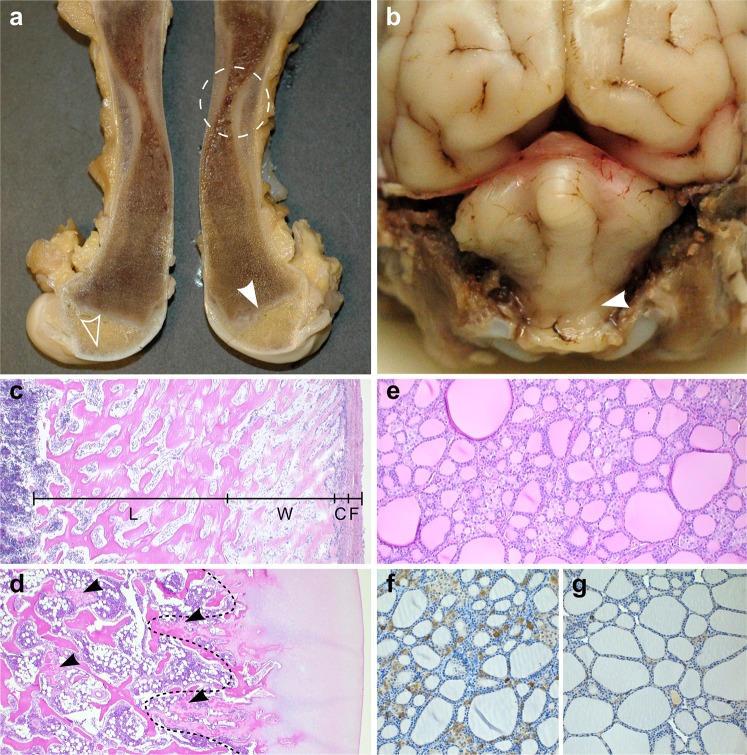
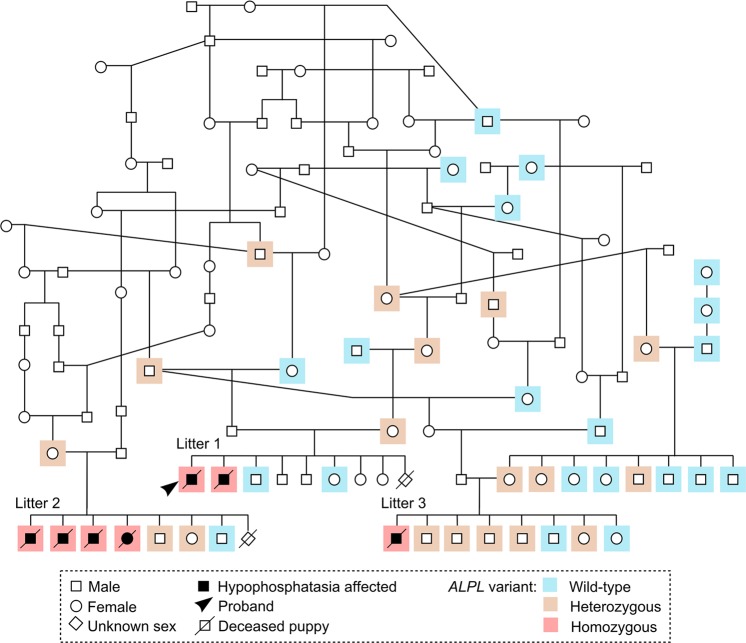
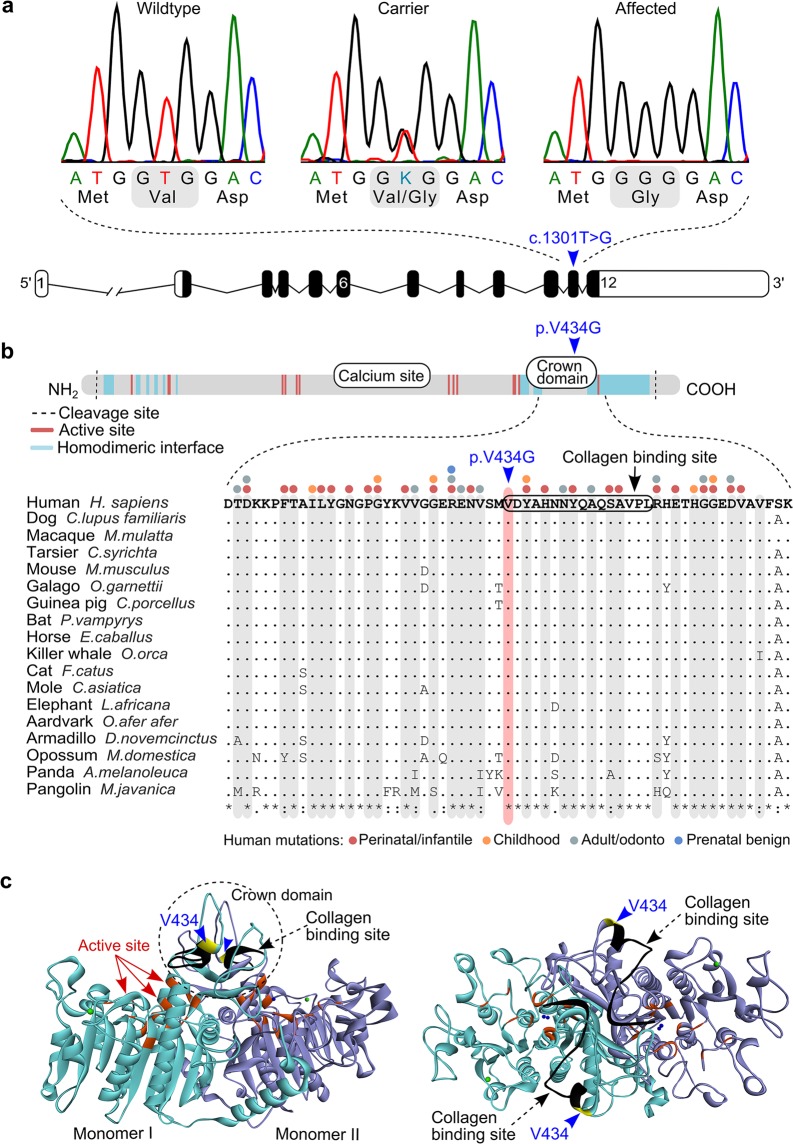
Inherited skeletal disorders affect both humans and animals. In the current study, we have performed series of clinical, pathological and genetic examinations to characterize a previously unreported skeletal disease in the Karelian Bear Dog (KBD) breed. The disease was recognized in seven KBD puppies with a variable presentation of skeletal hypomineralization, growth retardation, seizures and movement difficulties. Exome sequencing of one affected dog revealed a homozygous missense variant (c.1301T > G; p.V434G) in the tissue non-specific alkaline phosphatase gene, ALPL. The identified recessive variant showed full segregation with the disease in a cohort of 509 KBDs with a carrier frequency of 0.17 and was absent from 303 dogs from control breeds. In humans, recessive and dominant ALPL mutations cause hypophosphatasia (HPP), a metabolic bone disease with highly heterogeneous clinical manifestations, ranging from lethal perinatal hypomineralization to a relatively mild dental disease. Our study reports the first naturally occurring HPP in animals, resembling the human infantile form. The canine HPP model may serve as a preclinical model while a genetic test will assist in breeding programs.
Conflict of interest statement
During the study, H.L. was a shareholder of Genoscoper Laboratories Oy (until 12/2017), which offers genetic screening for the identified variant. H.L. continues to provide consultancy to the company. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
