

Cardiomyocyte substructure reverts to an immature phenotype during heart failure
- PMID: 30707448
- PMCID: PMC6441900
- DOI: 10.1113/JP277273
Cardiomyocyte substructure reverts to an immature phenotype during heart failure
Abstract
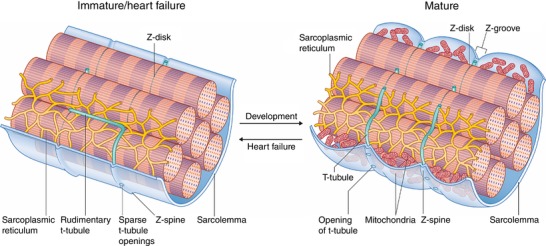
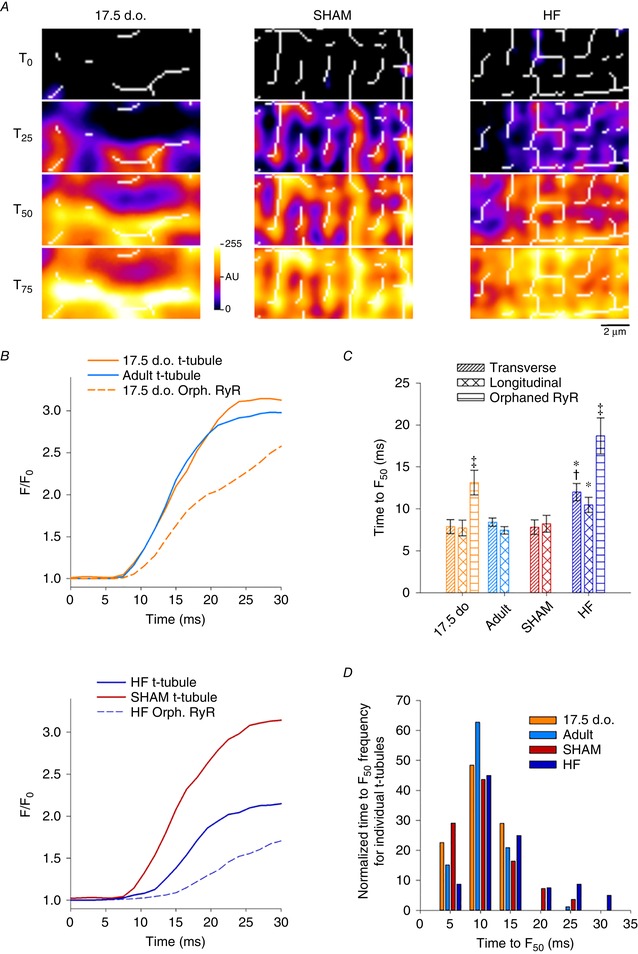
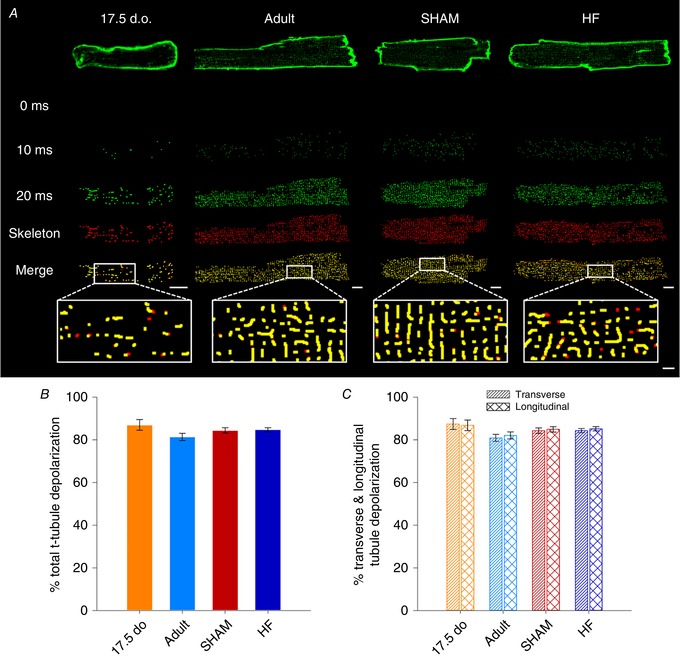
Key points: As reactivation of the fetal gene program has been implicated in pathological remodelling during heart failure (HF), we examined whether cardiomyocyte subcellular structure and function revert to an immature phenotype during this disease. Surface and internal membrane structures appeared gradually during development, and returned to a juvenile state during HF. Similarly, dyadic junctions between the cell membrane and sarcoplasmic reticulum were progressively 'packed' with L-type Ca2+ channels and ryanodine receptors during development, and 'unpacked' during HF. Despite similarities in subcellular structure, dyads were observed to be functional from early developmental stages, but exhibited an impaired ability to release Ca2+ in failing cardiomyocytes. Thus, while immature and failing cardiomyocytes share similarities in subcellular structure, these do not fully account for the marked impairment of Ca2+ homeostasis observed in HF.
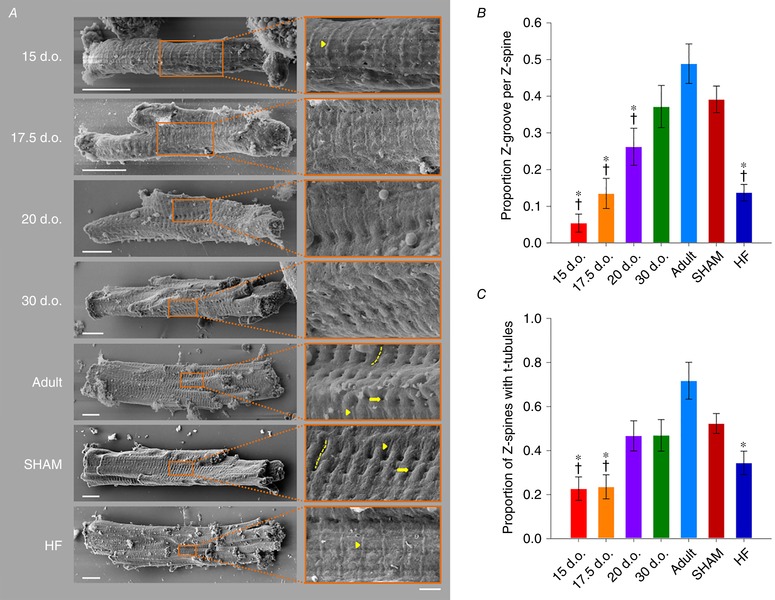
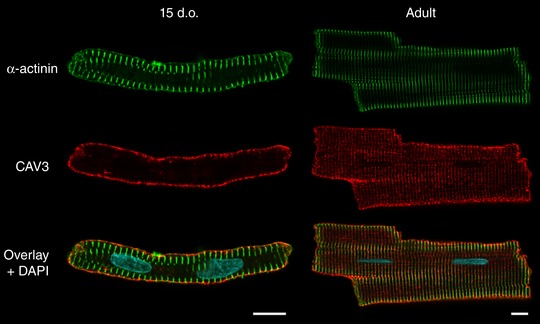
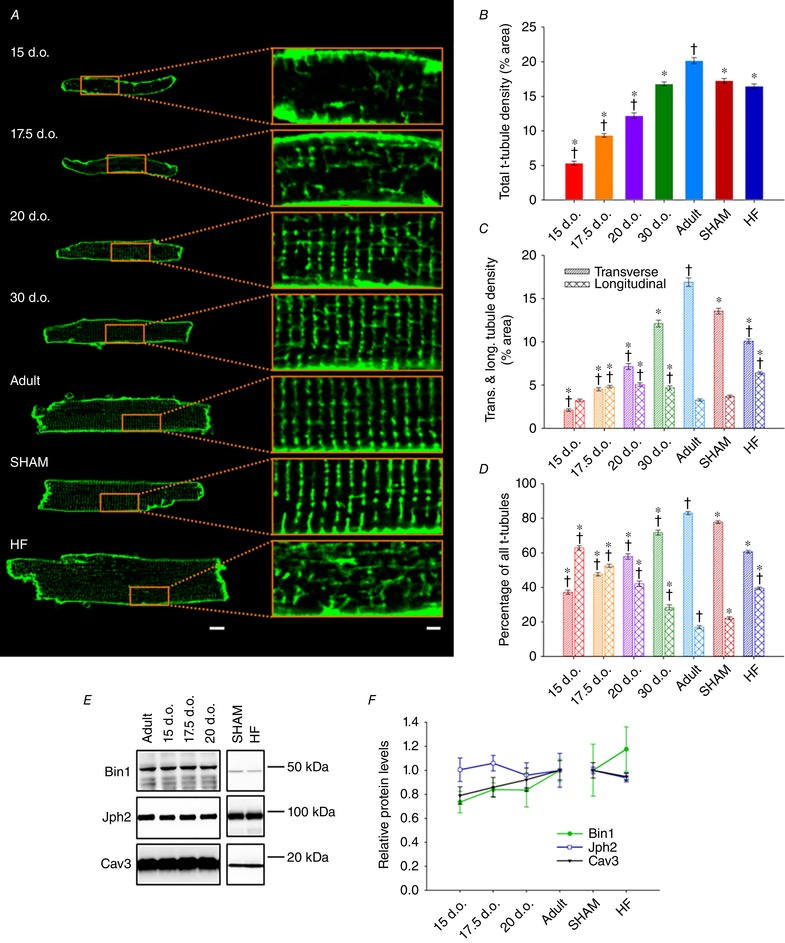
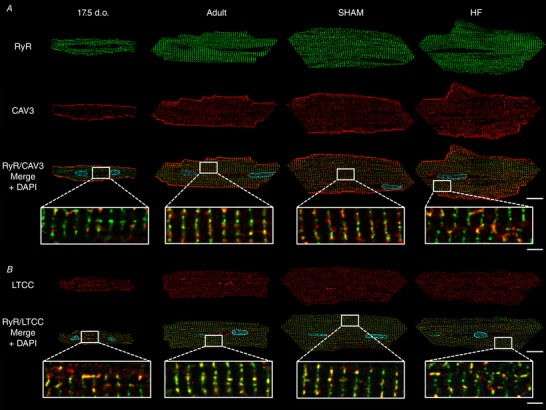
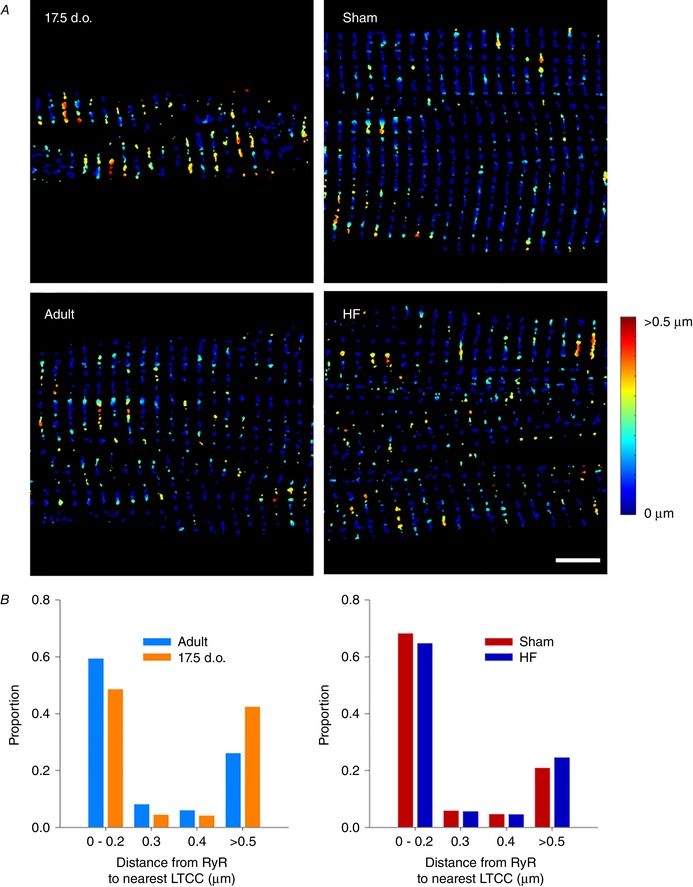
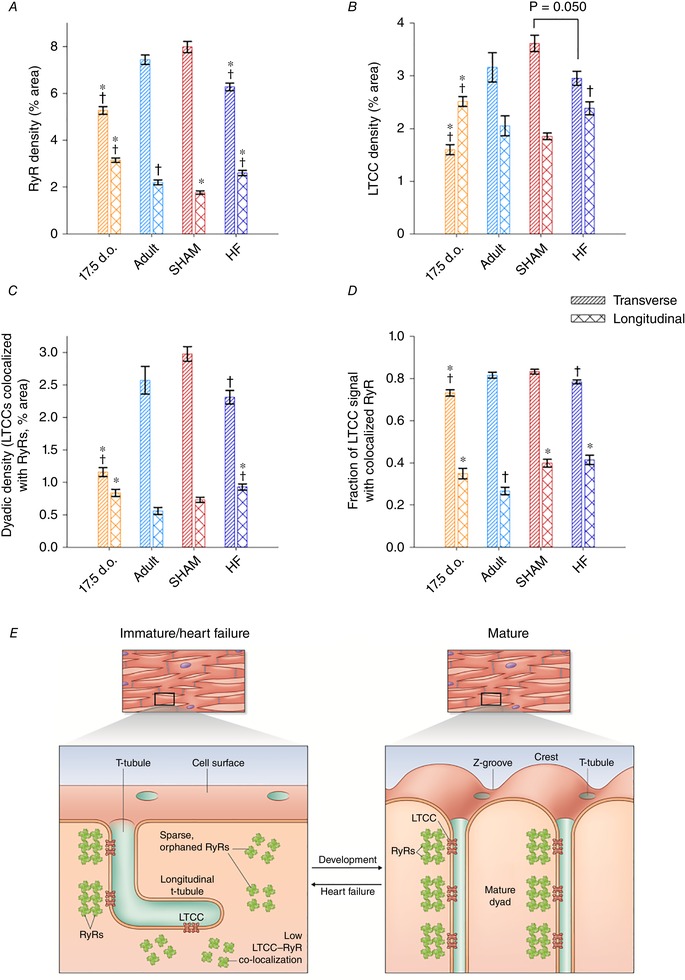
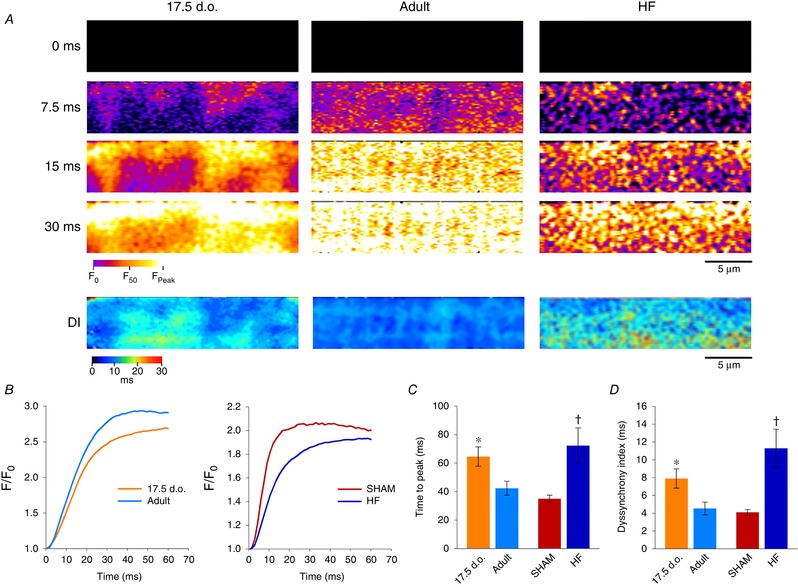
Abstract: Reactivation of the fetal gene programme has been implicated as a driver of pathological cardiac remodelling. Here we examined whether pathological remodelling of cardiomyocyte substructure and function during heart failure (HF) reflects a reversion to an immature phenotype. Using scanning electron microscopy, we observed that Z-grooves and t-tubule openings at the cell surface appeared gradually during cardiac development, and disappeared during HF. Confocal and super-resolution imaging within the cell interior revealed similar structural parallels; disorganization of t-tubules in failing cells was strikingly reminiscent of the late stages of postnatal development, with fewer transverse elements and a high proportion of longitudinal tubules. Ryanodine receptors (RyRs) were observed to be laid down in advance of developing t-tubules and similarly 'orphaned' in HF, although RyR distribution along Z-lines was relatively sparse. Indeed, nanoscale imaging revealed coordinated packing of L-type Ca2+ channels and RyRs into dyadic junctions during development, and orderly unpacking during HF. These findings support a 'last in, first out' paradigm, as the latest stages of dyadic structural development are reversed during disease. Paired imaging of t-tubules and Ca2+ showed that the disorganized arrangement of dyads in immature and failing cells promoted desynchronized and slowed Ca2+ release in these two states. However, while developing cells exhibited efficient triggering of Ca2+ release at newly formed dyads, dyadic function was impaired in failing cells despite similar organization of Ca2+ handling proteins. Thus, pathologically deficient Ca2+ homeostasis during HF is only partly linked to the re-emergence of immature subcellular structure, and additionally reflects lost dyadic functionality.
Keywords: calcium homeostasis; development; dyad; heart failure; t-tubule.
© 2019 The Authors. The Journal of Physiology published by John Wiley & Sons Ltd on behalf of The Physiological Society.
Figures
Comment in
-
Letter to the Editor.J Physiol. 2019 Jun;597(11):2965-2966. doi: 10.1113/JP278018. Epub 2019 Apr 14. J Physiol. 2019. PMID: 30924149 No abstract available.
-
Reply from M. Frisk, D. B. Lipsett and W. E. Louch.J Physiol. 2019 Jun;597(11):2967-2968. doi: 10.1113/JP278067. Epub 2019 May 12. J Physiol. 2019. PMID: 31021407 No abstract available.
References
-
- Aronsen JM, Espe EK, Skardal K, Hasic A, Zhang L & Sjaastad I (2017). Non‐invasive stratification of postinfarction rats based on degree of cardiac dysfunction using magnetic resonance imaging and echocardiography. Am J Physiol Heart Circ Physiol 312, H932–H942. - PubMed
-
- Bers DM (2002). Cardiac excitation–contraction coupling. Nature 415, 198–205. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous