

Short-term gain, long-term pain: the senescence life cycle and cancer
- PMID: 30709901
- PMCID: PMC6362810
- DOI: 10.1101/gad.320937.118
Short-term gain, long-term pain: the senescence life cycle and cancer
Abstract
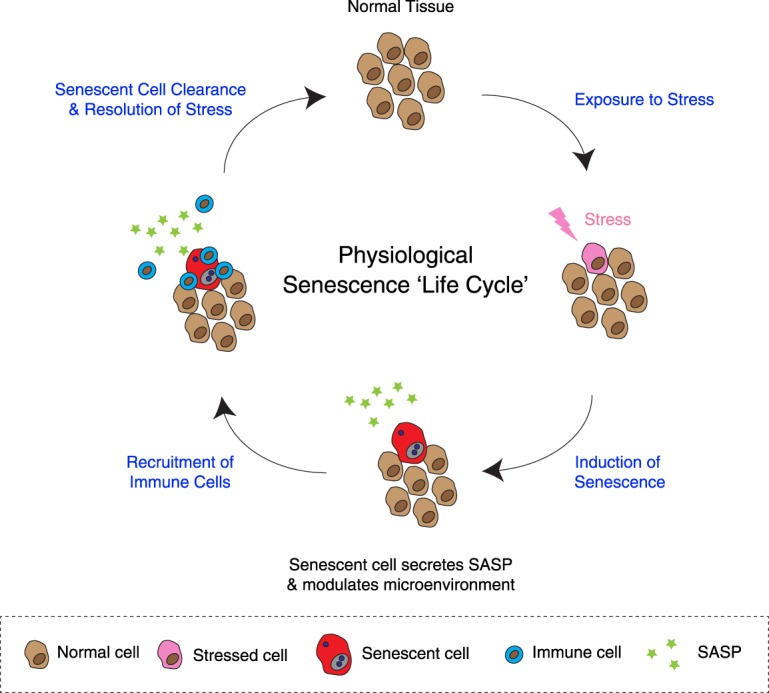
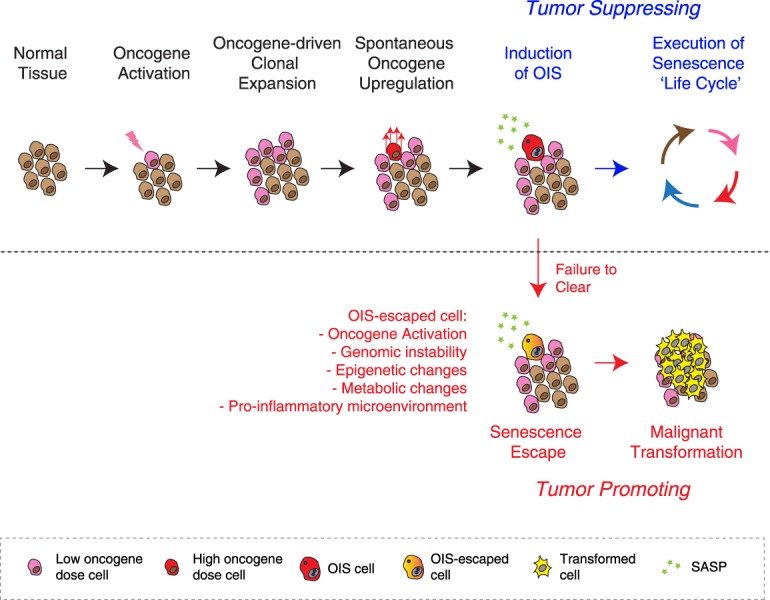
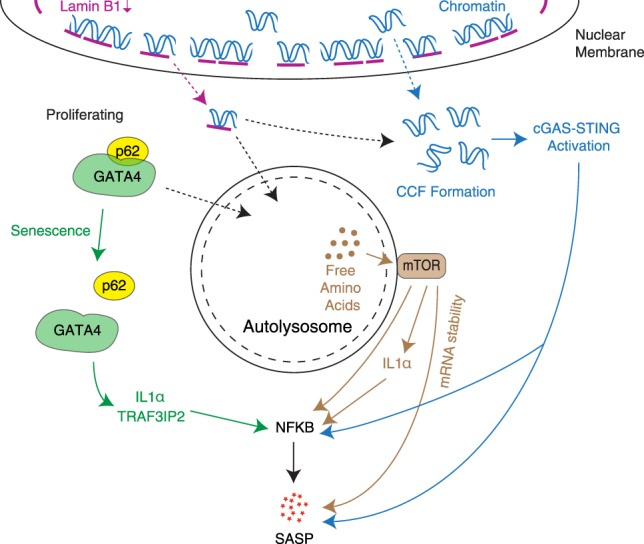
Originally thought of as a stress response end point, the view of cellular senescence has since evolved into one encompassing a wide range of physiological and pathological functions, including both protumorignic and antitumorigenic features. It has also become evident that senescence is a highly dynamic and heterogenous process. Efforts to reconcile the beneficial and detrimental features of senescence suggest that physiological functions require the transient presence of senescent cells in the tissue microenvironment. Here, we propose the concept of a physiological "senescence life cycle," which has pathological consequences if not executed in its entirety.
Keywords: cancer; epigenetics; inflammation; senescence.
© 2019 Chan and Narita; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources