

The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A
- PMID: 30709905
- PMCID: PMC6442034
- DOI: 10.1074/jbc.RA118.005008
The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A
Abstract
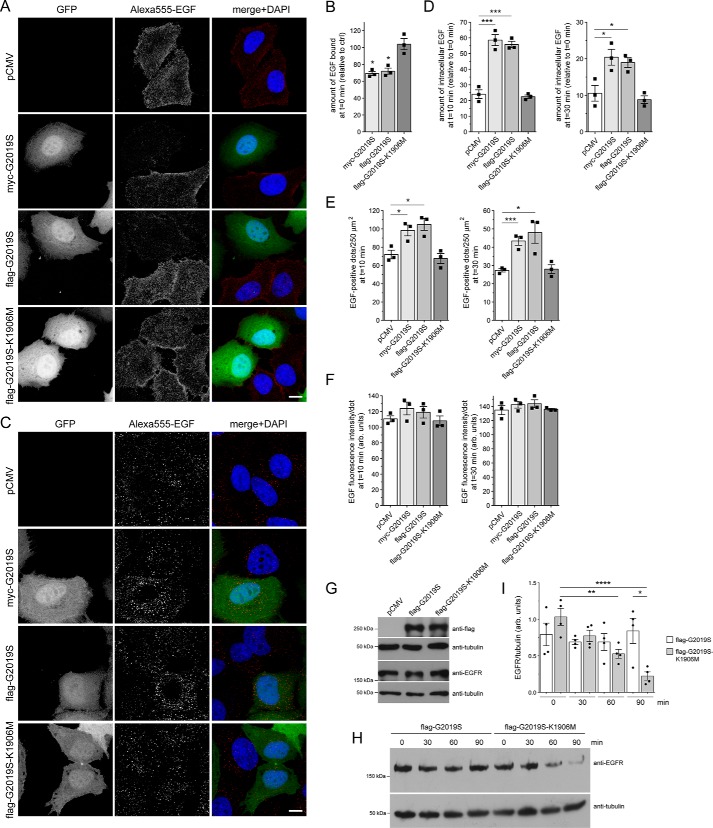
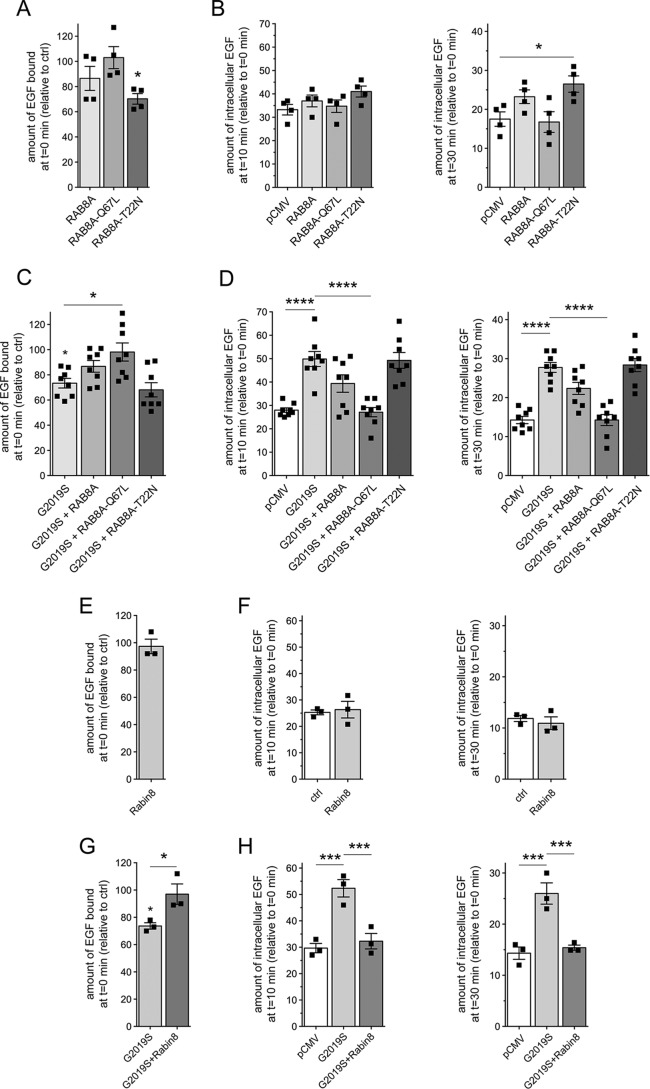
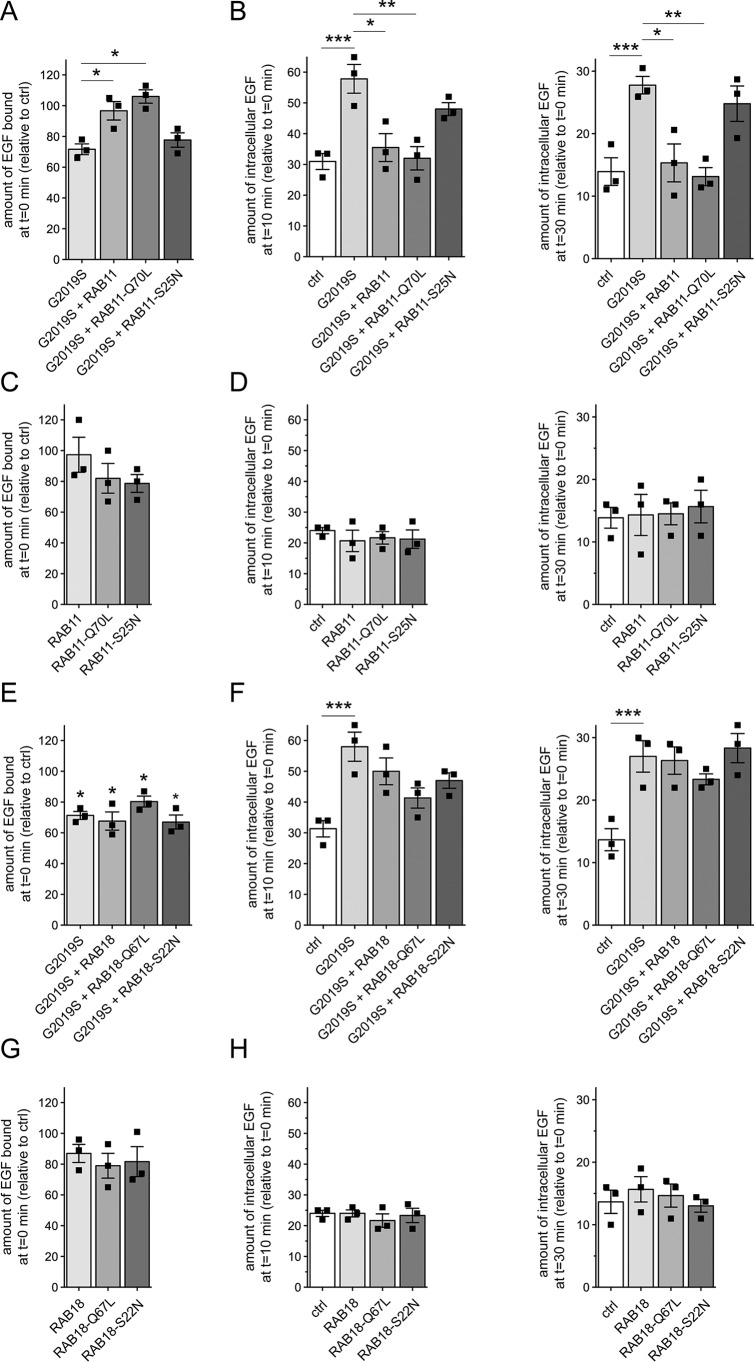
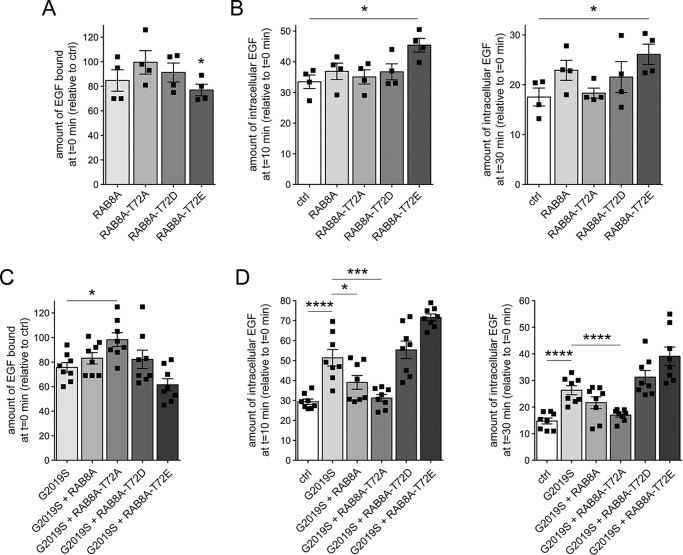
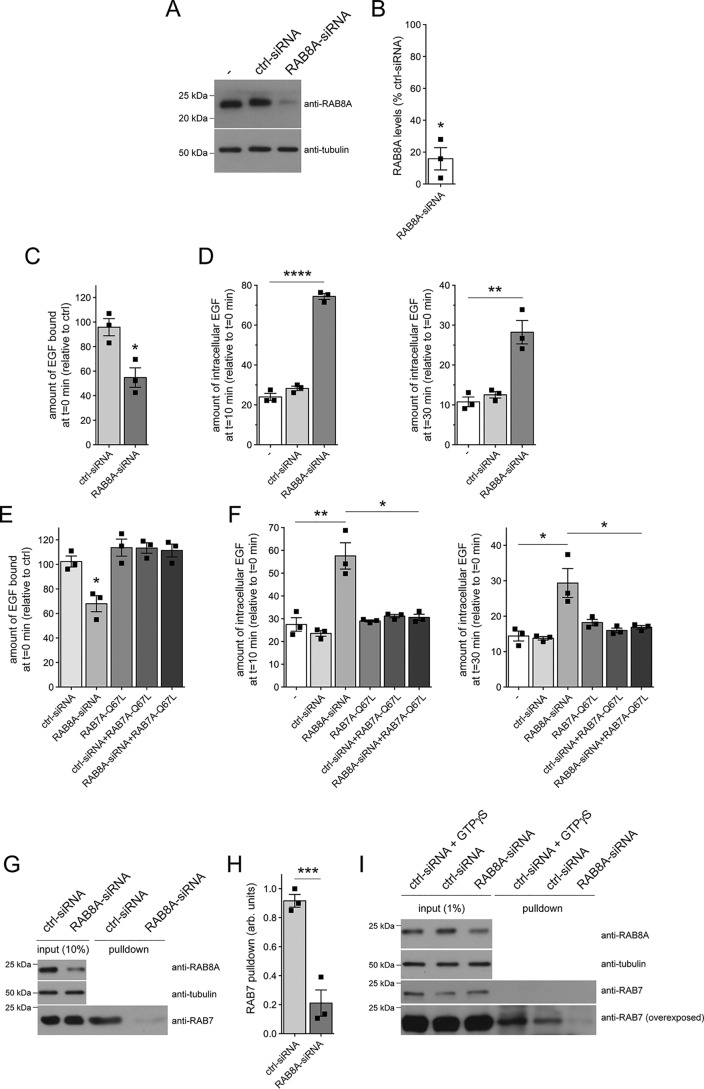
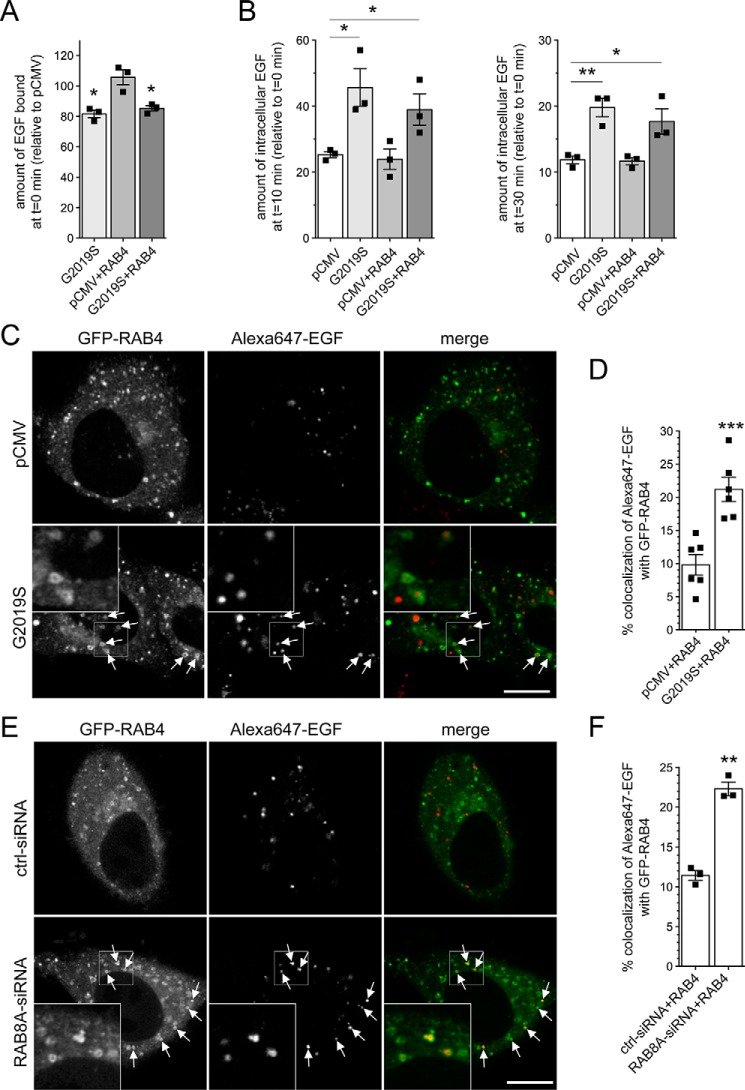
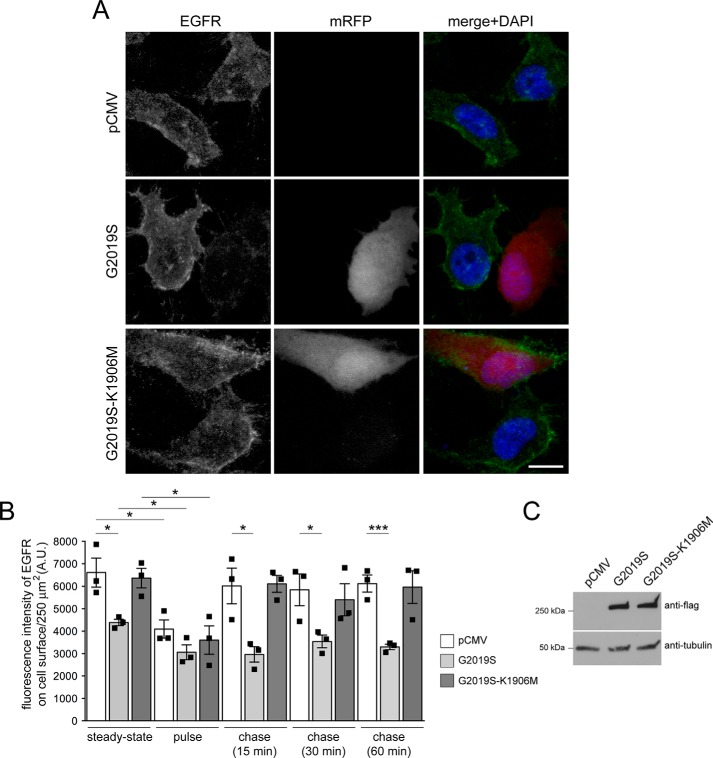
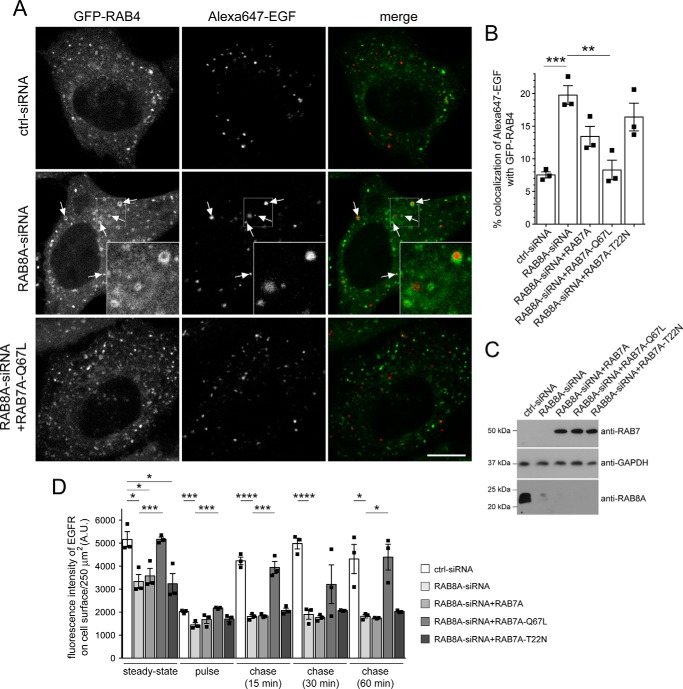
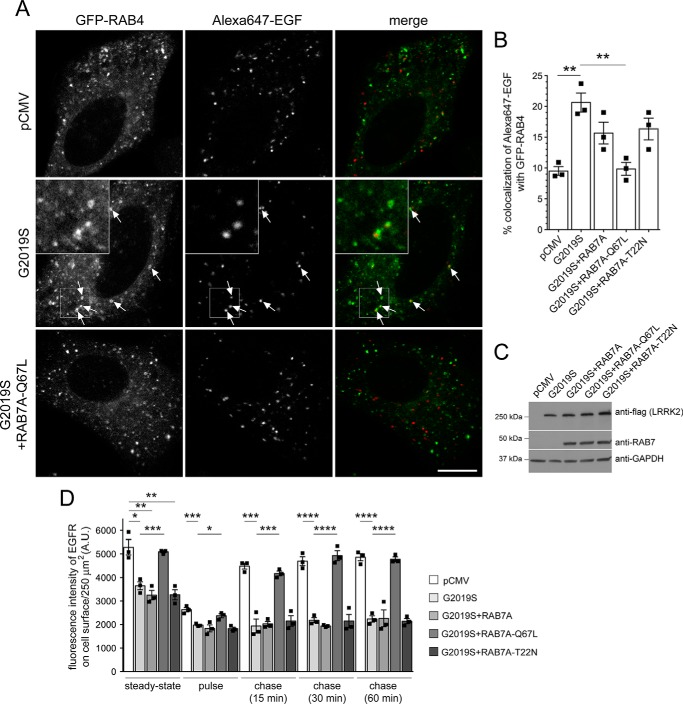
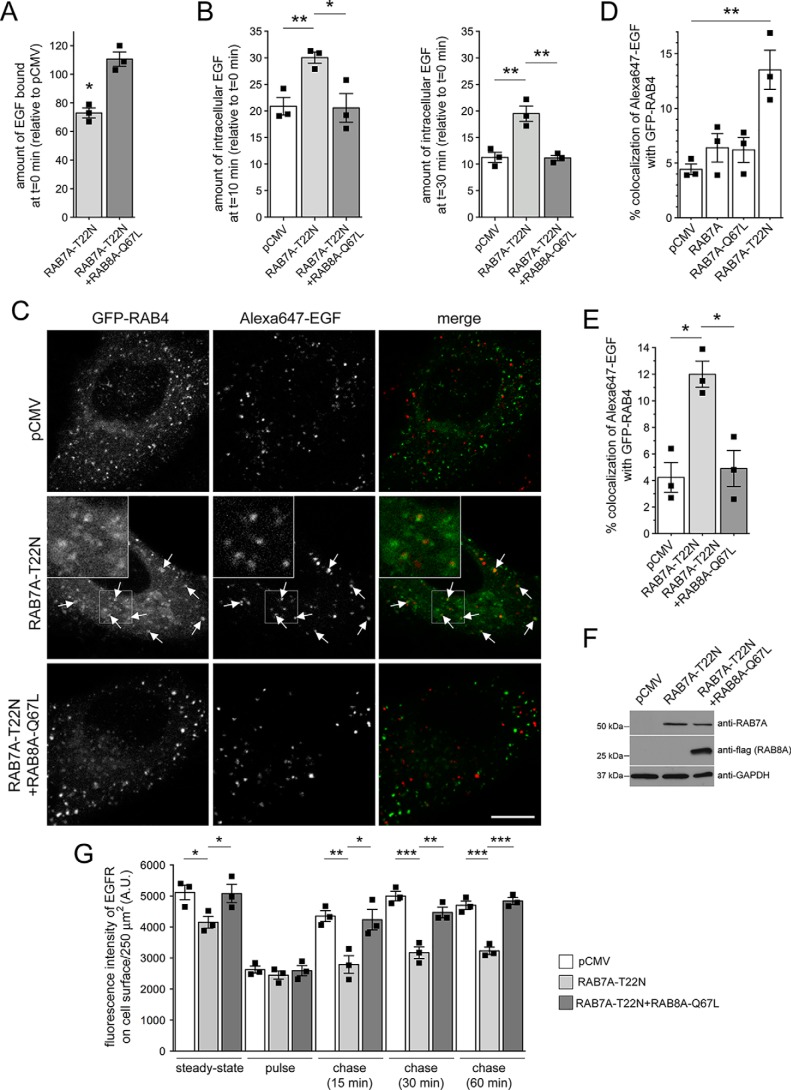
Mutations in the gene encoding for leucine-rich repeat kinase 2 (LRRK2) are a common cause of hereditary Parkinson's disease. LRRK2 regulates various intracellular vesicular trafficking pathways, including endolysosomal degradative events such as epidermal growth factor receptor (EGFR) degradation. Recent studies have revealed that a subset of RAB proteins involved in secretory and endocytic recycling are LRRK2 kinase substrates in vivo However, the effects of LRRK2-mediated phosphorylation of these substrates on membrane trafficking remain unknown. Here, using an array of immunofluorescence and pulldown assays, we report that expression of active or phosphodeficient RAB8A variants rescues the G2019S LRRK2-mediated effects on endolysosomal membrane trafficking. Similarly, up-regulation of the RAB11-Rabin8-RAB8A cascade, which activates RAB8A, also reverted these trafficking deficits. Loss of RAB8A mimicked the effects of G2019S LRRK2 on endolysosomal trafficking and decreased RAB7A activity. Expression of pathogenic G2019S LRRK2 or loss of RAB8A interfered with EGFR degradation by causing its accumulation in a RAB4-positive endocytic compartment, which was accompanied by a deficit in EGFR recycling and was rescued upon expression of active RAB7A. Dominant-negative RAB7A expression resulted in similar deficits in EGF degradation, accumulation in a RAB4 compartment, and deficits in EGFR recycling, which were all rescued upon expression of active RAB8A. Taken together, these findings suggest that, by impairing RAB8A function, pathogenic G2019S LRRK2 deregulates endolysosomal transport and endocytic recycling events.
Keywords: GTPase; Parkinson disease; RAB7A; RAB8A; Rab; early recycling compartment; endolysosome; leucine-rich repeat kinase 2 (LRRK2); lysosome; neurodegeneration; protein homeostasis; receptor endocytosis; receptor recycling.
© 2019 Rivero-Ríos et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
Similar articles
-
Distinct Roles for RAB10 and RAB29 in Pathogenic LRRK2-Mediated Endolysosomal Trafficking Alterations.Cells. 2020 Jul 17;9(7):1719. doi: 10.3390/cells9071719. Cells. 2020. PMID: 32709066 Free PMC article.
-
LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity.Hum Mol Genet. 2014 Dec 20;23(25):6779-96. doi: 10.1093/hmg/ddu395. Epub 2014 Jul 30. Hum Mol Genet. 2014. PMID: 25080504
-
Mutations in LRRK2 linked to Parkinson disease sequester Rab8a to damaged lysosomes and regulate transferrin-mediated iron uptake in microglia.PLoS Biol. 2021 Dec 16;19(12):e3001480. doi: 10.1371/journal.pbio.3001480. eCollection 2021 Dec. PLoS Biol. 2021. PMID: 34914695 Free PMC article.
-
LRRK2 and Rab GTPases.Biochem Soc Trans. 2018 Dec 17;46(6):1707-1712. doi: 10.1042/BST20180470. Epub 2018 Nov 22. Biochem Soc Trans. 2018. PMID: 30467121 Review.
-
An Update on the Interplay between LRRK2, Rab GTPases and Parkinson's Disease.Biomolecules. 2023 Nov 13;13(11):1645. doi: 10.3390/biom13111645. Biomolecules. 2023. PMID: 38002327 Free PMC article. Review.
Cited by
-
PRKAA2, MTOR, and TFEB in the regulation of lysosomal damage response and autophagy.J Mol Med (Berl). 2024 Mar;102(3):287-311. doi: 10.1007/s00109-023-02411-7. Epub 2024 Jan 6. J Mol Med (Berl). 2024. PMID: 38183492 Review.
-
The LRRK2 signaling network converges on a centriolar phospho-Rab10/RILPL1 complex to cause deficits in centrosome cohesion and cell polarization.Biol Open. 2022 Aug 15;11(8):bio059468. doi: 10.1242/bio.059468. Epub 2022 Jul 29. Biol Open. 2022. PMID: 35776681 Free PMC article.
-
Sequential screening nominates the Parkinson's disease associated kinase LRRK2 as a regulator of Clathrin-mediated endocytosis.Neurobiol Dis. 2020 Jul;141:104948. doi: 10.1016/j.nbd.2020.104948. Epub 2020 May 17. Neurobiol Dis. 2020. PMID: 32434048 Free PMC article.
-
Brain Iron Metabolism, Redox Balance and Neurological Diseases.Antioxidants (Basel). 2023 Jun 16;12(6):1289. doi: 10.3390/antiox12061289. Antioxidants (Basel). 2023. PMID: 37372019 Free PMC article. Review.
-
LRRK2 and the Endolysosomal System in Parkinson's Disease.J Parkinsons Dis. 2020;10(4):1271-1291. doi: 10.3233/JPD-202138. J Parkinsons Dis. 2020. PMID: 33044192 Free PMC article. Review.
References
-
- Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Carrera I. M., Pena A. S., et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600 10.1016/j.neuron.2004.10.023 - DOI - PubMed
-
- Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., et al. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 10.1016/j.neuron.2004.11.005 - DOI - PubMed
-
- Healy D. G., Falchi M., O'Sullivan S. S., Bonifati V., Durr A., Bressman S., Brice A., Aasly J., Zabetian C. P., Goldwurm S., Ferreira J. J., Tolosa E., Kay D. M., Klein C., Williams D. R., et al. (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 7, 583–590 10.1016/S1474-4422(08)70117-0 - DOI - PMC - PubMed
-
- Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M. P., Beilina A., Blackinton J., Thomas K. J., Ahmad R., Miller D. W., Kesavapany S., Singleton A., Lees A., et al. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341 10.1016/j.nbd.2006.04.001 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
