

Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute
- PMID: 30710114
- PMCID: PMC6862721
- DOI: 10.1038/s41596-018-0113-7
Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute
Abstract
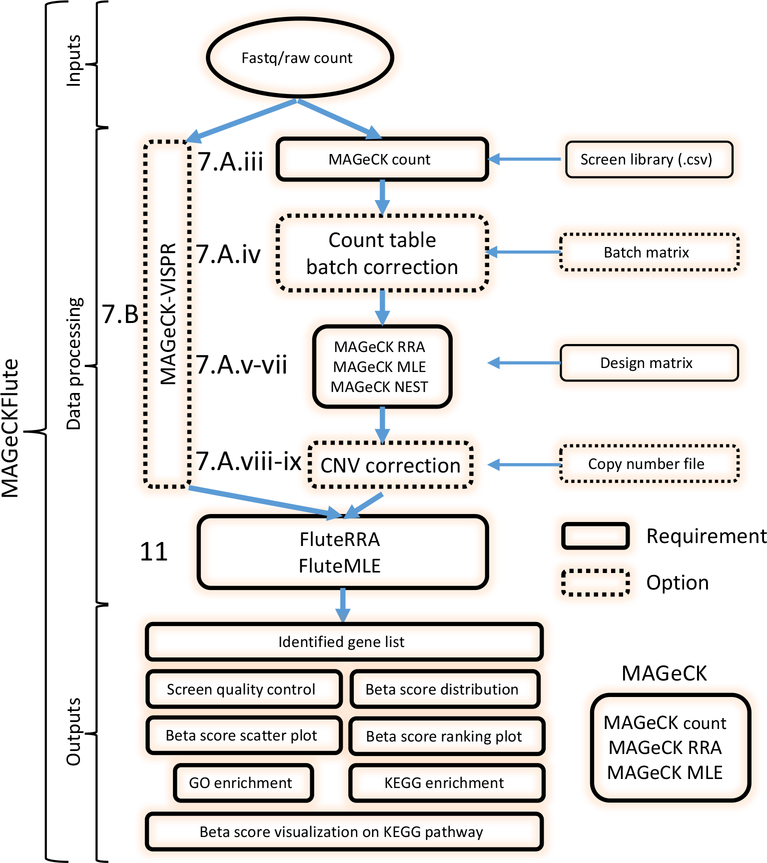
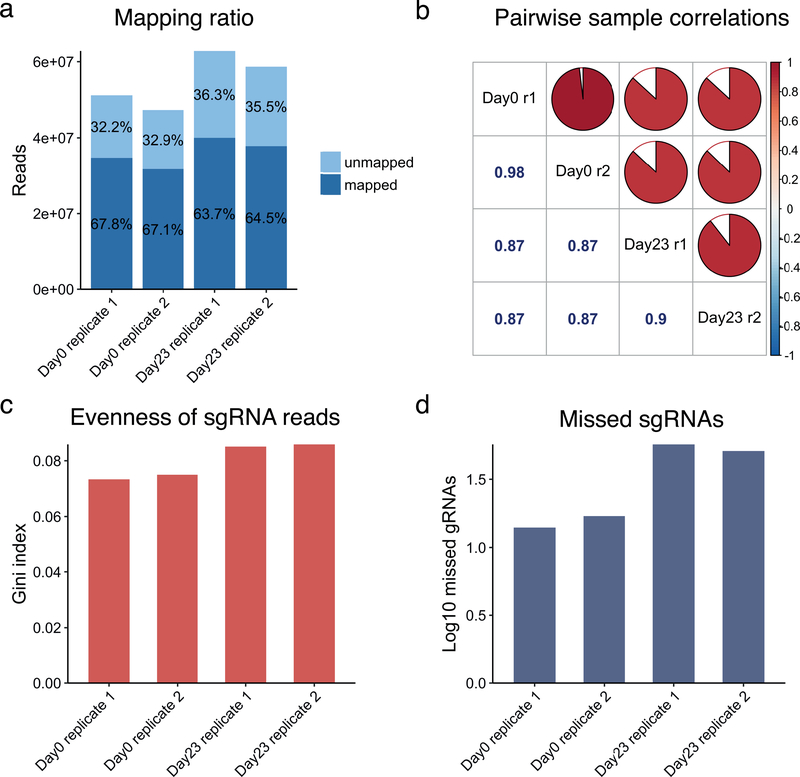
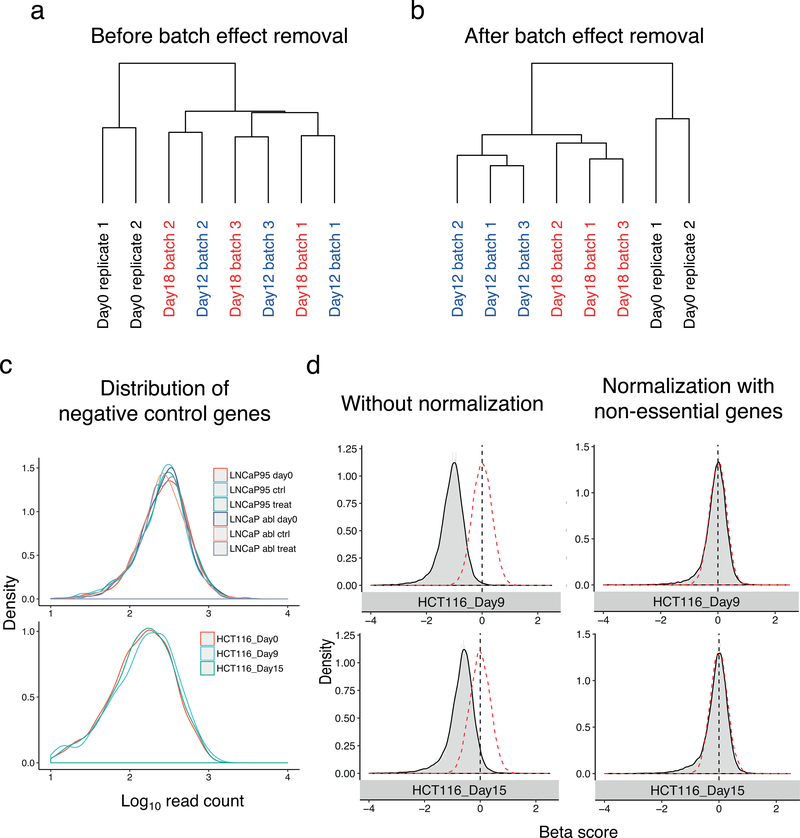
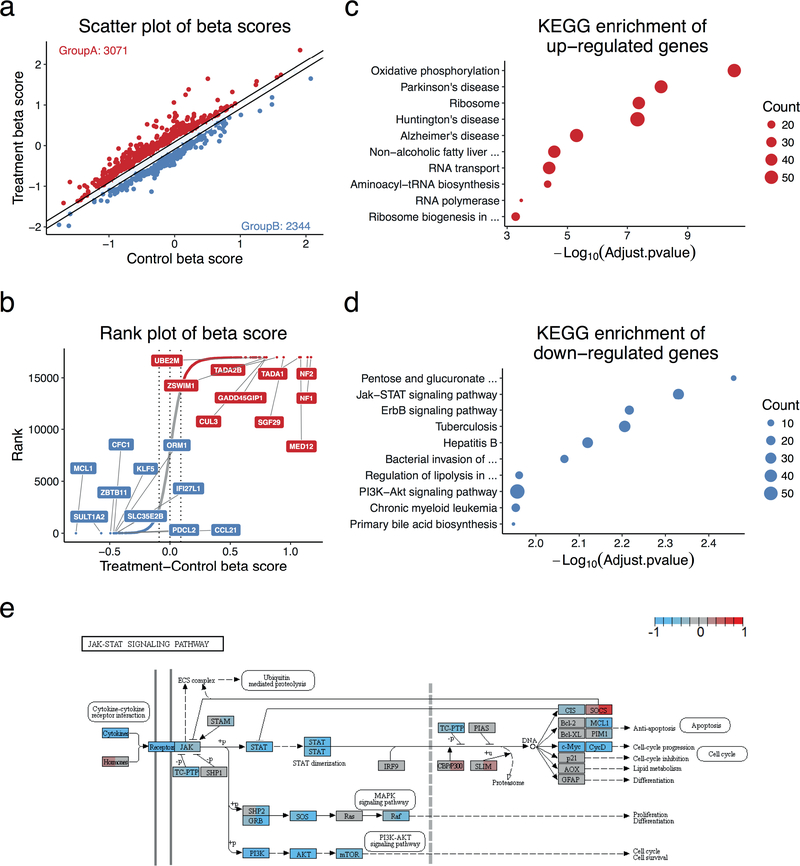
Genome-wide screening using CRISPR coupled with nuclease Cas9 (CRISPR-Cas9) is a powerful technology for the systematic evaluation of gene function. Statistically principled analysis is needed for the accurate identification of gene hits and associated pathways. Here, we describe how to perform computational analysis of CRISPR screens using the MAGeCKFlute pipeline. MAGeCKFlute combines the MAGeCK and MAGeCK-VISPR algorithms and incorporates additional downstream analysis functionalities. MAGeCKFlute is distinguished from other currently available tools by its comprehensive pipeline, which contains a series of functions for analyzing CRISPR screen data. This protocol explains how to use MAGeCKFlute to perform quality control (QC), normalization, batch effect removal, copy-number bias correction, gene hit identification and downstream functional enrichment analysis for CRISPR screens. We also describe gene identification and data analysis in CRISPR screens involving drug treatment. Completing the entire MAGeCKFlute pipeline requires ~3 h on a desktop computer running Linux or Mac OS with R support.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
